第 16 章 基督教打破家庭观念
令欧洲退出亲戚关系的是宗教,不是政治;对欧洲家庭性质的普遍误会;天主教会摧毁延伸的亲戚团体;英国个人主义甚至在欧洲也属极端
我迄今所叙述的世界三个地区,其国家制度都自部落社会脱颖而出。中国、印度和中东的早期社会组织,都以父系家族的血统为基础,建立国家是为了克服部落社会的局限。每一个案例中,建国者想方设法让个人忠于国家,而不是忠于地方上的亲族团体。以领土和中央合法统治权力为基础的制度,不得不重叠在顽固的分支式社会之上。最极端的对策来自阿拉伯和奥斯曼帝国,他们绑架儿童,使之在人造家庭中长大,只忠于国家,不忠于自己的亲戚。
但在这些案例中,不让亲戚关系成为社会组织基础的建国努力,自上而下,都归于失败。事实上,这些社会的制度发展历史,大多涉及亲族团体的重新问政——我称之为家族制复辟。所以,秦朝和西汉所创建的非人格化国家制度,在东汉崩溃时又落到强大宗族手中,这些家庭继续成为中国政坛中的重要角色,直到隋唐。印度在创建强大的非人格化制度上,一开始就成绩平平,以分支式迦提组织起来的印度村庄,其社会生活大体上又与这些制度毫不相干。土耳其国家是最为成功的,在小亚细亚和巴尔干半岛的心脏地区削弱了部落组织的影响,但在治理不严的阿拉伯省却不如人意。事实上,奥斯曼帝国在边远的贝都因(Bedouin)社区,仅行使非常有限的统治,其部落组织至今保持原样。所有这些地区——中国、印度、中东——家庭和亲戚团体至今仍然强大,成为社会组织和身份的来源,远远超过欧洲或北美。在中国台湾和南方地区尚有成熟的分支世系家族,印度婚姻仍是家庭而不是个人的结合。部落的依附关系在阿拉伯中东无所不在,尤其是在贝都因的群体中。
例外的欧洲
欧洲的亲戚关系采纳不同形式。人口统计学家约翰·哈吉那尔(John Hajnal)在 1965 年的文章中注意到,西欧婚姻模式与世界上几乎任何其他地方形成强烈对照。①西欧男女倾向于晚婚,从总体上讲,不结婚的比率较高,这两个因素导致相对较低的出生率;更多年轻女子参加工作,家庭中有更多平等,由于晚婚,女子又有较多机会获取财产。这不仅是当代现象,哈吉那尔把这种模式的时期定在 1400 年到 1650 年。
西欧与世界其他地方的其他差异也很突出。共同祖先的亲戚团体所组成的社区,其在欧洲的消失远远早于哈吉那尔所指出的。对欧洲人而言,亲戚和后裔很重要,特别是国王和贵族,他们有实质性的经济资源传给子孙。但跟中国贵族不同,他们没有陷入表亲的专横,因为分割遗产和长子继承权的原则早已深入人心。在中世纪,欧洲人享有更多自由,无须征得大批亲戚的同意,便可任意处置自己的土地和动产。
换言之,欧洲社会很早就是个人主义的。在婚姻、财产和其他私人事务上,当家做主的是个人,而不是家庭或亲戚团体。家庭中的个人主义是所有其他个人主义的基础。个人主义无须等待国家的出现,无须等待它来宣告个人法律权利,并行使强制权力来予以保障。更确切地说,个人已在享受实质性的自由,无须承担对亲戚的社会义务,先有这样的社会,再来建起国家。在欧洲,社会发展走在政治发展的前列。
欧洲何时退出亲戚关系?如果不是政治,转型动力何在?前者的答案是:蹂躏罗马帝国的日耳曼部落,在皈依基督教后不久,就开始退出。后者的答案是:天主教会。
马克思的错误
很明显,现代欧洲人的祖先都曾组成部落。他们的亲戚关系、法律、习惯、宗教实践,只要能找到的,19 世纪伟大的历史人类学家都已作了详细记载,如甫斯特尔·德·库朗日、亨利·梅因②、弗雷德里克·波洛克(Frederick Pollock)、弗雷德里克·梅特兰(Frederic Maitland)③、保罗·维诺格拉多夫(Paul Vinogradoff)。他们是比较人类学家,掌握不同文化的渊博知识,为父系亲戚组织之间的相似而感到吃惊。那些组织分布于世界各地,如印度、希腊、日耳曼的社会。④
19 世纪的历史人类学家相信,亲戚组织随着时间的推移而进化,人类社会有普遍的发展模式,从亲戚团体的大集团,转向个别男女自愿结合的小家庭。梅因有个著名概念:现代化涉及从“身份到契约”的过渡。⑤换言之,早期社会将社会地位赋予个人,安排一切,从婚配、职业到宗教信仰。相比之下,现代社会的个人可随意与人签约,走进不同社会关系,其中最重要的是婚姻合同。但梅因没有提出一种动态理论,以解说过渡是何时和如何发生的。
实际上,对欧洲亲戚模式的过渡时间和过渡原因存在很多误解。很多人相信,跟世界上的其他民族类似,欧洲人始终居住于部落或庞大的家庭团体,一直到工业革命。其时,机器生产的压力和社会流动的必要性,才将之打破。根据这个见解,工业化带来经济变化和核心小家庭出现,都属于这同一过程。⑥
这个见解很可能来自早期现代化理论。卡尔·马克思在《共产党宣言》中宣称,资产阶级“撕下了罩在家庭关系上的温情脉脉的面纱,把这种关系变成了纯粹的金钱关系”。促使资产阶级兴起的,依次是技术革新和物质生产方式的变化。马克斯·韦伯指出,传统社会和现代社会之间有严重断裂。传统社会的特征是:广泛的亲戚关系,宗教或亲戚的约束对市场交易设限,缺乏个人社会流动性,基于传统、宗教、超凡魅力的非正式社会规范。而现代社会是个人主义的、平等的、以优秀和市场为导向的、流动的,并以法理型合法性权威组建起来。韦伯主张,这些特征属于一个整体,如果由教士指定价格,或财产受亲戚义务束缚,这样社会就不能发展出高效的市场经济。他相信,这种理性的现代化仅在西方出现,并把向现代化的过渡定在 16 世纪和 17 世纪的一系列事件,包括宗教改革(Protestant Reformation)和启蒙运动(Enlightenment)。所以,马克思主义者倾向于认为,经济变化促使个人主义和核心小家庭的兴起,而韦伯则把基督新教当作主要动力。总之,依他们看,这个变化仅有几百年历史。
从身份到契约
20 世纪的社会历史学家和人类学家,把从身份到契约的过渡一直往前提。我已提及,哈吉那尔认为欧洲的特殊模式始于 15 世纪和 16 世纪。艾伦·麦克法兰(Alan MacFarlane)对英国个人主义起源的研究显示,生前任意处置财产和死后在遗嘱中剥夺子女继承权,早在 16 世纪初就获得英国普通法的承认。⑦这很重要,因为他所标志的“农民社会”中,如东欧和世界大部分区域,亲戚义务大大限制了业主出售土地的能力。农民社会的特征就是大家庭,产权要么共有,要么陷于亲戚的相互依赖之中。这样的社会中,许多非经济因素把农民牢牢绑在他们所耕种的土地上,诸如祖先葬于此之类的理由。
但麦克法兰注意到,土地所有权(seisin)流行于英国,至少还要往前再提三个世纪。根据一项研究,15 世纪晚期英国某区的地产转户中,生前赠与家人的占 15%,死后遗赠给家人的占 10%。⑧更早的是 12 世纪末 13 世纪初,英国的佃户(villeins,不得随意离开土地)无须获得领主的许可,已在购买、出售、出租土地。⑨
如要衡量复杂亲戚组织的衰退,就要考量女子拥有和处置财产的法律权利。父系家族的社会中,女子嫁与宗族中的男子,或给宗族生下男性后裔,方才取得法律地位。寡妇和未婚女儿有分享遗产的权利,但通常必须将宗族的财产留在父系家族中。1066 年的诺曼征服(Norman Conquest)之后不久,英国女子就可自由拥有和处置财产,并可将之卖给外人。至少从 13 世纪起,她们不但可拥有土地和动产,而且可起诉他人,或被他人起诉,甚至可签署遗嘱和合同,无须征得男子监护人的许可。父系社会一旦承认这种权利,就会破坏宗族控制财产的能力,从而破坏社会制度的整体。⑩所以,女子拥有和遗赠财产的能力是部落组织退化的标志。它显示,严格的父系社会规则已经消失。
根据麦克法兰,早期英国个人主义的一个有趣标志是“扶养合同”。它最早出现于 13 世纪,由孩子和父母签署。共同祖先的后裔团体所组成的部落社会,通常崇拜共同祖先。儒家道德的大部分涉及孩子照料父母的义务,尤其是儿子。儒家道德家讲得很清楚,对父母的义务大于对自己孩子的,中国法律严惩不孝子女。
英国的习俗却不同,父母活着时,如把产权愚蠢地转移给孩子,就得不到惯例的剩余权利。中世纪有一首诗歌,描述了父亲将财产移交给儿子的故事:儿子后来觉得扶养父亲的负担太重,便开始施以虐待。一天,父亲冷得直打寒战,儿子叫孙子送去一只麻布袋,“小男孩把麻布袋一割为二,一半留给爷爷,另一半带回给父亲。他的意思是,现在父亲虐待爷爷,等到自己长大,也会如法炮制,给他半个麻布袋以御寒”。⑪为了避免如此的困境,父母与孩子签署扶养合同,规定孩子在继承父母财产后所承担的扶养责任。“贝德福德的一对夫妇在 1294 年放弃财产,作为回报,将得到食物、饮料、主屋的居住;如果两对夫妇发生争吵,老夫妇会搬到另外房子,将在圣米迦勒节(Michaelmas)获得五十六蒲式耳的谷物,其中二十四蒲式耳的小麦,十二蒲式耳的大麦,十二蒲式耳的大豆和豌豆,八蒲式耳的燕麦。此外,他们还将得到这另外房子的一切,可动的和固定的。”⑫
让马克思暴跳如雷的“纯粹的金钱关系”,似乎不是 18 世纪资产阶级的发明,其在英国的出现比资产阶级的兴起早了好多世纪。将父母寄放在疗养院,在西欧有很深的历史根源。这显示,与马克思的主张恰恰相反,资本主义只是社会关系和习俗变化的后果,而不是原因。
如果说欧洲在 13 世纪离开复杂的亲戚关系,即从身份过渡到契约,这依然太迟。伟大的法国历史学家马克·布洛赫注意到,封建主义在 9 至 10 世纪兴起之前,亲戚关系是社会组织的基础。部落宗族之间的血亲复仇在欧洲社会有悠久历史,我们对此很熟悉,只要看看莎士比亚的《罗密欧与朱丽叶》就知道了。此外,布洛赫证明,在那段时期,亲戚团体或庞大家族共同拥有财产,即使个人已开始随意处置土地,卖主仍需获得亲戚团体的同意。⑬
不过,布洛赫提示,可以追溯到像中国、印度、中东那样单一祖先的巨大父系宗族,很久以前就在欧洲消失了。“罗马家族视男性后裔为绝对重要,立场异常坚定。但此事到了封建时代,已变成闻所未闻。”作为证据,他指出,中世纪的欧洲人从不单凭父亲来追溯他们的后裔;而在部落社会中,为了维持宗族分支的界线,这是不可或缺的。在整个中世纪时期,母亲让女儿冠母姓是很普遍的,这在中国那样的父系社会是不可想象的。个人经常认为自己属于两个相互平等的家庭,母亲的和父亲的。两个杰出家庭的子孙往往合并两个宗族的姓氏(如瓦勒里·季斯卡·德斯坦 Valéry Giscard d’Estaing,其中季斯卡和德斯坦都是姓氏[编按:瓦勒里·季斯卡·德斯坦系法国前总统,1974—1981 年任职]。今日西班牙人喜用父母的双姓)。到 13 世纪,类似当代的核心家庭已在欧洲遍地开花。血亲复仇很难继续,因为报仇圆圈变得越来越小,很多人觉得,自己与争论双方都有关联。⑭
在布洛赫看来,某种意义上,封建主义的整个制度可被理解为迫不得已的调整,这是为了适应社会上的隔绝,因为亲戚关系不再是社会团结的来源。自 7 世纪晚期起,欧洲遭受了一系列外国侵略者的蹂躏:来自北方的维京人、来自南方借道于北非和西班牙的阿拉伯人或撒拉森人(Saracens)、来自东方的匈牙利人。即使阿拉伯人受挫于图尔战役,穆斯林对地中海的控制仍然切断欧洲与拜占庭和北非的贸易,它曾是罗马经济的基础。⑮随着卡洛林帝国(Carolingian)在 9 世纪的式微,城市也开始凋零,受无数军阀骚扰的居民撤回自给自足的村庄。
在这欧洲文明的最低点,由于更大政治结构的倒塌,亲戚关系试图卷土重来。但其时,欧洲的父系宗族结构已变得如此脆弱,以致不能成为社会支持的来源。封建主义兴起,成为亲戚关系的替代:
暴力气氛所孕育的无数危险,时时都在威胁个人。甚至在封建的初期,亲戚团体似乎不能提供足够的保护。根据它们当时存在的形式,这些团体的范围太模糊,太多变。父母都可界定后裔这种二元性,更造成了深刻的破坏。这就是为什么,人们被迫寻求或接纳其他的纽带。在这点上,历史是决定性的。仍有强大父系团体的地区——北海边上的日耳曼地区和英伦岛上的凯尔特地区——对属臣、采邑、庄园一无所知。亲戚关系只是封建社会的必要元素之一,它的相对孱弱解释了封建主义的出现。⑯
封建主义是指,个人自愿屈服于无亲戚关系的他人,仅仅是以服务交换保护。“国家和家庭不再提供足够的保护,村庄的社区仅能维持界线之内的秩序,城市的社区几乎不存在。各处软弱者觉得有必要获得强人的庇护,而强人必须通过说服或强制来获得签约下属的支持,以保障自己的威望、财富、人身安全。”⑰
但我们还没算出欧洲脱离亲戚关系的日期,以及合适的因果关系。⑱社会人类学家杰克·古迪(Jack Goody),为过渡日期作出了最令人信服的解释。他把过渡的起点提至 6 世纪,将责任放在基督教身上——具体地说,放在天主教会的机构利益上。⑲
古迪注意到,罗马帝国结束时,与众不同的西欧婚姻模式从主要的地中海模式分化出来。包括罗马家族的地中海模式,属于严格的父系家族或父系社会,具有分支式的社会组织。父系团体倾向于同族通婚,有些更偏爱交叉表亲的婚姻。(我在第 11 章提及,交叉表亲的婚姻在印度南部的达罗毗荼文化中非常流行,在阿拉伯世界、普什图人[Pashtuns]、库尔德人、众多突厥人中也很普遍。)男女有严格的分隔,女子拥有财产或参与公共事务的机会很少。在所有这些方面,西欧的模式是截然不同的:分配遗产时男女都有份、禁止交叉表亲的婚姻、提倡异族通婚、女子有更多的产权和参与权。
天主教会促动了这一分化,它极力反对四种行为:与近亲结婚、与兄弟的寡妇结婚(levirate,即所谓的兄终弟及或夫兄弟婚)、领养孩子、离婚。教皇格里高利一世在 6 世纪敦使异教的盎格鲁—萨克逊人皈依基督教,尊敬的比德(Venerable Bede)在报告此事时就提及,格里高利直率谴责部落实行的与近亲和兄弟的寡妇的婚姻。后来的教堂法令禁止纳妾,提倡一生不分的一夫一妻婚姻。⑳
古迪认为,这些禁令并不直接依据《圣经》或基督教经典。被禁的行为在耶稣诞生的巴勒斯坦是很普遍的,耶稣父母可能就是交叉表亲的婚姻,与兄弟的寡妇结婚在犹太人中也很流行。事实上,基督教福音是反家庭的:在《马太福音》中,耶稣说,“爱父母超过爱我的人,不配我;爱子女超过爱我的人,也不配我”。古迪又称,这些话语来自宣称耶稣将统治尘世一千年的先知,他试图招募人们离开安全的亲戚团体,进入新兴的分裂教派。赞成禁令的神学观点则经常来自《旧约》,犹太人对此却有不同见解。
根据古迪,教会坚持这个立场的原因,与其说为了神学,倒不如说为了教堂自己的物质利益。交叉表亲的婚姻(或任何其他近亲的婚姻)、与兄弟的寡妇结婚、纳妾、领养孩子、离婚都是他所谓的“继承策略”;借此,亲戚团体得以继续控制代代相传的财产。其时,欧洲和地中海世界的居民寿命低于 35 岁。夫妇生下儿子、长到成人、再一次传宗接代的可能性相当低。因此,为了让人们得以孕育继承人,社会提供各式合法途径。讨论中国时,已解说过纳妾一事。在一夫一妻的社会,离婚可被视作变相纳妾。哥哥在生孩子之前就已去世,嫂子就与弟弟结婚,以确保哥哥的财产将与弟弟的融合在一起。交叉表亲的婚姻能保证家产留在自家人的手中。无论什么情形,教会有计划地切断将财产传给后裔的各种途径。同时,它又强烈提倡信徒向教会自愿捐出土地和财产。拥有财产但无继承人的基督徒日益增多,得益的便是教会。㉑
西欧女子相对较高的地位也是教会追求自身利益的意外结果。寡妇若在家庭团体内重新结婚,会将财产归还部落。教会尽量使之难以实现,所以她本人必须拥有财产。女子有权拥有和处置自己的财产,对教会大有裨益,无子女寡妇和老处女变成了捐献的一大来源。女子有权拥有财产破坏了单传原则,从而敲响了父系宗族的丧钟。㉒
规则发生变化后的数世纪中,天主教会在财政上非常成功,这绝对不是牵强附会。7 世纪结束之前,法国富饶土地的三分之一都在教会手中;从 8 世纪到 9 世纪,在法国北部、日耳曼、意大利的教会财产翻了一番。㉓这些捐献使教会成为一个在经济和政治上都很强大的机构,为格里高利七世的叙任权斗争(investiture conflict)铺平道路(见后文第 18 章)。这些捐献,跟富裕穆斯林给伊斯兰慈善事业瓦克夫的捐赠有相似之处,但后者主要是富人避税和遗赠子女的对策。而在天主教的欧洲,无子女寡妇和老处女所捐出的土地,则没有附带任何条件。教会因此发现自己成了大地主,在欧洲各地管理庄园,监督农奴的经济生产。这帮助教会履行其赈济饥民和照顾病人的使命,使教士阶层和男女修道院的大幅扩充成为可能,也使内部规则和等级制度的发展变得必不可少。这一切让教会在中世纪的政治舞台中成为一名独立角色。
这些变化对西欧的部落组织构成相当大的破坏。日耳曼、挪威、马札尔(Magyar)、斯拉夫的部落皈依基督教后,仅在两代或三代的时间就见证了其亲戚架构的解散。事实上,这种皈依植根于政治,如马札尔国王伊斯特万(István,或 St. Stephen)在 1000 年接受圣餐。但社会风俗和家庭规则中的实质性变化,不靠政治当局,而靠运作于社会和文化层次的教会。
欧洲建国的社会背景
欧洲(以及其殖民地)是个例外,因为它脱离复杂的亲戚关系,首先在社会和文化层次,而不在政治层次。在某种意义上,教会采取政治行为,更改了婚姻和遗产的规则,但其动机却是经济的。教会不是其所在领土的主权统治者,更确切地说,只是一个社会参与者,它的影响只在制定文化规则。因此,中世纪时,欧洲社会已经非常个人主义了。它早于欧洲国家建设的开端,比宗教改革、启蒙运动、工业革命更早了数个世纪。家庭中的变化,与其说是这些现代化巨变的结果,倒不如说是促进现代化发生的有利条件。16 世纪在意大利、英国、荷兰兴起的资本主义,不必去克服印度和中国那样的亲戚大集团的抵抗,后者亟欲保护自己拥有的实质性财产。相反,资本主义在那些社会顺利扎根,它们已有私人产权的传统,财产经常在陌生人之间转手。
这不是说,欧洲的建国者一帆风顺,没有遇上既得利益的社会建制。恰恰相反,我们在第 21 章继续讲述欧洲国家起源时,将看到各式强大的社会参与者,他们在创建法治和负责制政府方面至关重要。虽然没有氏族或部落,但有既得利益的贵族,他们在封建时期积累下了财富、军队、法律地位。
这些社会建制是封建的,并不基于亲戚关系。这一事实,对后世的欧洲政治发展来说,造成了重大差别。属臣的封建关系是强者和弱者自愿签署的合同,规定了双方的法律义务。它将高度不平等和等级化的社会形式化,但也为个人主义(签署合同的是个人而不是亲戚团体)和法律人的理解树立先例。历史学家杰诺·苏克斯(Jenö Szücs)认为,地主与农民之间的关系到 1200 年便获得契约特征,从而打下了将尊严扩充到农民阶层的基础。自那以后,“西方每一次农民反抗,都在表述地主违反合同所激怒的尊严,都在诉求自己的‘自由’权利”。㉔但这种事没有发生于下列社会:土地产权以亲戚关系和惯例为基础的,或以某亲戚团体称王称霸于另一亲戚团体为基础的。
以封建制度替代亲戚关系建制,对地方治理的功效而言,另有重要的政治影响。宗族和封建制度都在不同时期发挥主权和统治的功能,尤其是在中央国家式微时。它们都可提供地方安全、司法和经济生活的组织。但封建制度更为灵活,因为依据的是合同,更能组织决定性的集体行动,因为其等级分明。跟宗族中的权威不同,封建领主的权利一旦获得合法确认,便不需要持续的重新谈判。财产的法律文件,无论在强者或弱者的手中,都代表自由买卖的权利,不受基于亲戚的社会制度的限制。地方上的领主可“代表”社区讲话,但部落领袖做不到。如我们所知,欧洲殖民者在印度和非洲经常犯的错误,就是假设部落领袖相当于封建社会的地方领主。在事实上,两者截然不同。
马克斯·韦伯的遗产之一,就是以价值概念来考量宗教对政治和经济的影响。他的新教工作伦理(work ethic),据说通过工作的神圣化,而直接影响工业革命中企业家的行为。价值肯定是重要的,上帝之下人人平等的基督原则,使女子更容易获得拥有财产的平等权利。
但此类解释经常引申新的疑问,为何有些宗教价值首先在社会中获得提倡,并深入人心。教会攻击延展的亲戚关系,就是一例,这些价值并不起源于基督教原则。毕竟,同是基督徒的君士坦丁堡东正教,并没有设法改变婚姻和遗产的法律。所以,紧密相关的亲戚社区在拜占庭统治的地区存活很久。例如,塞尔维亚代代相传的著名乡村团体“杂住盖”(zadruga),以长期血亲复仇著称的阿尔巴尼亚氏族。这些建制消失于西欧,归功于教会的物质利益和权力。教会对社会价值的控制,变成了为己谋利的工具。从这个角度看,经济龟站在宗教龟的上面;但从另一角度看,宗教龟又站在更为底下的经济龟上。 不管其动机是宗教的,还是经济的,天主教会变成了独立的政治参与者,其建制化的程度,远远超过其他社会的宗教权力。中国从没发展出超越祖先崇拜或鬼神崇拜的本土宗教。相比之下,宗教发明一开始就塑造了印度和穆斯林世界,成为政治权力的重要制衡。但在伊斯兰教逊尼派的世界,以及印度次大陆,宗教权力从没凝聚成国家之外的中央官僚机构。它只在欧洲出现,与现代欧洲国家的发展和今天所谓的法治的出现又密不可分。
第三部分 法治
第 17 章 法治的起源
法律在早期国家形成中的作用凸显欧洲的例外;法治的定义和争论;法律优先于立法的哈耶克理论;英国普通法依据皇家权力来加强国家的合法性
欧洲的政治发展是个例外。欧洲社会得以较早脱离部落组织,却没有依靠自上而下的政治权力。欧洲例外还表现在,其早期建国者的杰出能力,与其说是在军事上,倒不如说在分配正义上。欧洲国家权力和合法性的增长,与法治的涌现密不可分。
早期欧洲国家分配的只是正义,不一定是法律。法律植根于他处,或在宗教(像上一章所讨论的有关婚姻和家庭的法令),或在部落和其他社区的习俗。早期欧洲国家偶尔立法——即制定新法律——但其权力和合法性更多依赖公正执法,所执的法无须是自己订出的。
弄清法律和立法之间的差异,对理解法治是至关重要的。似乎有多少法律学者,就会有多少“法治”的定义,很像“民主”这个字眼。①我所使用的,符合思考此一现象的西方重要潮流:法律是凝聚社区的有关正义的一组抽象规则。在前现代社会,制定法律的权威据信是超凡的,或神权,或古老习俗,或自然。②另一方面,立法属于现在所谓的制定法(positive law),它在发挥政治权力的功能。就像国王、男爵、总统、立法院、军阀,凭借自己的权力和权威,在制定和执行新的规则。如果有高于任何立法的现存法律,方能说有法治的存在。这意味着,拥有政治权力的个人必须接受法律的束缚。这不是说,立法机构不可制定新法,它们如想在法治中发挥作用,必须依据现存法律的规则来制定新法,不可随心所欲。
法律的最初理解,即制定者是神权、古老习俗或自然,指的是人们不得更改法律,但可以为特殊情境作出妥善解释,有时还是必须的。现代时期,随着宗教权威的走低,自然法信徒的锐减,我们开始将法律视作人造的东西,但必须经过严格程序,以确保它符合基本规则的广泛共识。法律和立法之间的差异,现在相当于宪法和一般法律之间的差异。前者具有更严格的要求,例如绝大多数人的投票同意。在当代美国,这表示国会通过的新法律,必须符合现存的更为重要的宪法,一切以最高法院的解释为准。
迄今为止,我讨论了政治发展中的建国以及国家集中和使用权力的能力。法治是政治秩序中的另一组件,以限制国家权力。对行政权力的最初制衡不是民主集会或选举,而是人们坚信统治者必须依法行事。所以,国家建设和法治一直在紧张氛围中共存。一方面,统治者在法律范围内行事,或以法律的名义行事,这可提高自己的权威。另一方面,法律可防止他们做随心所欲的事,不能只考虑私人利益,还要考虑整个共同体的利益。所以,政治权力的欲望经常威胁法治,从 17 世纪避开议会自筹税收的英国国王,到 20 世纪以法外行刑队来对付恐怖主义的拉丁美洲政府,皆是如此。
法治的现代迷惑
在当代发展中国家,最大政治缺点之一就是法治的相对软弱。当代国家的所有组件中,高效法律机构也许是最难构建的。军事和征税的机构,天然来自人类基本的掠夺本能。军阀组织民兵向社区榨取资源,这并不困难。在另一极端,民主选举的安排也相对容易(只是比较昂贵),何况今天还有国际组织的援助。③但法律机构必须遍布整个国家,持续不断,长期运作。它们需要设施,投资于律师、法官及法庭其他职员的训练,还有最终执法的警察。但最重要的,法律机构必须被视作合法和权威的,不仅在普通人眼中,而且在更有力的精英眼中。做到这一点,证明是颇不容易的。今天,拉丁美洲绝大多数国家是民主的,但其法治却非常软弱,到处是收贿的警官和逃税的法官。俄罗斯联邦仍举行民主选举,自总统而下的精英都违法乱纪,肆无忌惮,尤其是在弗拉基米尔·普京当政之后。
有很多文献,将法治的建立与经济发展挂起钩来。④这些文献从根本上反映出一条重要观点,即现代资本经济世界的涌现,在很大程度上归功于既存的法治,缺乏高效的法治是贫困国家不能取得较高增长的主要原因。
但这些文献非常混乱,在法治的基本定义和它的存在与否上,前后又不一致。此外,将法治的不同组件与经济增长挂钩的理论,其实证经验有点靠不住;将它投射到马尔萨斯经济条件下的社会,困惑只会加倍。我们在讲法治起源的历史之前,需要清除一下当代讨论所留下的累赘。
经济学家谈论法治时,通常指现代产权和合同执行。⑤现代产权是指个人拥有的财产,可自由买卖,不受亲戚团体、宗教当局、国家的限制。经济增长受产权和合同的影响,这理论非常直截了当。没人会做长期投资,除非知道自己的产权是安全的。如果政府突然对某种投资增税,像乌克兰在 20 世纪 90 年代签署移动电话基建协议后所作的,投资者可能会在中途改弦易辙,并对将来项目心灰意冷。同样,贸易需要法律机构来维持合同,裁判合同双方不可避免的争执。合同的规则越透明,合同的维持越公正,就会鼓励越多的贸易。这就是为何很多经济学家强调,“可信承诺”是国家制度发展的重要标志。
这个法治定义与本章开头的那个略有重叠。显而易见,如果政府觉得自己在各方面都享有主权,不受既存法治的束缚,那么无人可阻止它充公自己公民或外国贸易伙伴的财产。如果普遍的法律规则,一旦牵涉到强大的精英阶层或最强大的政府本身,就无法得到执行,那么产权或贸易的安全可能只是空头支票。政治学家巴里·温加斯特(Barry Weingast)注意到,强大的国家既可保护产权,也可取消产权。⑥
另一方面,有“足够好”的产权和合同执行,允许经济的发展,但没有真正的法治(即法律是至高无上的意思),这完全可行。⑦……中国经济取得三十多年的两位数增长,并不需要“法治”的抽象承诺。1978 年,共产党以包产到户的法律解散了人民公社,但没有恢复中国农民的现代产权(个人转让土地的权利)。更确切地说,他们只获得可遗传的土地使用权(长期租赁权),类似于奥斯曼帝国中央省的农民。这些权利已经“足够好”,导致农业产量在改革后的四年功夫翻了一番。
古代帝制中国没有法治。另一方面,正常时期的中华帝国很可能在地方层次享有“足够好”的产权,至少将农业生产效率提高到其时技术所容许的极限。那时的产权与今天中国农民的产权相比,不会相差很多,与其说受到掠夺性国家的约束,倒不如说受到亲戚关系的约束。父系宗族将无数的权利和习俗强加于财产之上,一直到 20 世纪的中华民国,家庭仍有权利限制土地的出售。⑧
此外,不是很清楚,最好的现代产权足以在实质上提高生产效率,还是足以在马尔萨斯式社会中创建出现代资本主义。确保技术持续进步的其他建制(如科学方法、大学、人力资源、研究实验室、鼓励探险和试验的文化氛围,等等)尚未问世时,单凭良好产权所创造的生产效率增长仍然有限,因此不能假设技术的持续进步。⑨
所以,经济学家对法治下现代产权和合同执行的强调,可能有两个错位。首先,在技术持续革新的当代世界,虽然没有至高无上的法治,“足够好”的产权仍足以创造高度的经济增长。其次,在马尔萨斯式世界中,即使有现代产权和法治的存在,还是无法取得如此的增长,因为限制增长的约束出自其他地方。
法治还有一个定义,对经济生活具有极大影响,不管是在前现代还是在当代。这就是人身安全,即从暴力的自然状态中退出,从事日常活动,不用担心被杀或被抢。它存在时,我们视之为理所当然;它缺席时,我们会尤其珍惜。
最终,谈论法治时一定要弄清法律对象,即是说,受法律保护的法人群体。社会的基本执法对大家是一视同仁的,但保护公民免受国家任意侵犯的法治,最初往往只适用于特权阶层的少数。换言之,法律仅仅保护靠近或控制国家的精英的利益。在此意义上,法律就像苏格拉底在柏拉图《理想国》中所标榜的“强盗帮派的正义。”
举塞维涅夫人(Mme. de Sévigné)写给女儿的信为例,她是 17 世纪法国最著名的沙龙赞助人之一。这位聪明敏感的女子描绘,士兵在布列塔尼征集新税,把老人和孩子从家中赶出,再在屋子里寻找可供夺取的财产。因为不付税,大约六十名市民将在下一天上绞刑架。她继续写道:“那个手舞足蹈、想偷印花税纸的闲汉在车轮上就刑,被割成四块,分别在城市四个角落示众。”⑩
显而易见,法国国家不会向塞维涅夫人和她朋友圈子施以如此激烈的惩罚。我们将在第 23 章看到,它将繁重税赋仅仅加给平民,因为它太尊重贵族的产权和私人安全。所以,说 17 世纪法国没有法治是不正确的,但法律并没认为平民也是法人,也享有与贵族相同的权利。美国初创时也是如此,否定非裔美国人、妇女、美洲原住民——除了拥有财产的白人男子——的选举权。民主化的过程逐渐拓展法治范围,以包纳所有的居民。
法治定义的混乱,其所造成的后果之一是富国设计的改善法治计划,很少在贫穷国家产生效果。⑪住在法治国家的幸运儿,往往不懂它如何首次涌现,误把法治的外表当作法治的实质。例如,“相互制衡”是强大法治社会的特征,政府各部门监督彼此的行为。但制衡的正式存在,并不等于强有力的民主统治。法庭可被用来阻挠集体行动,如当代印度,其冗长的司法上诉可拖死重要的基建项目。它又可被用来对抗政府的愿望,以保护精英利益。1905 年最高法院的洛克纳诉纽约州案(Lochner v. New York),其宗旨就是击败限制工时的立法,以保护企业利益。所以,分权的形式常常名不副实,与守法社会的主旨无法对应。
在接踵而来的讨论中,我们将从尽量广阔的角度去关注法治的发展:法律本身——整套正义规则——来自何方?产权、合同执行、商法的特定规则如何发展至今?最高政治当局如何接受法律的至高无上?
法律早于立法的哈耶克理论
伟大的奥地利经济学家弗里德里希·哈耶克,发展了关于法律起源的精深理论,为法治的涵义提供了重要见解,成为今日人们思考法律的框架。哈耶克被称作当代自由至上主义的教父,但自由至上主义者并不反对规则。根据哈耶克,“共同规则的存在使社会中个人的和平共处成为可能”。⑫在法律起源上,哈耶克把批判矛头指向所谓的“唯理主义”或“建构主义”理解。这种理解思路认为,立法者理性地研究社会问题,从而发明法律,以建立自以为更好的社会秩序。在哈耶克看来,建构主义只是过去三百年的自负,尤其是部分法国思想家,包括笛卡尔(Descartes)和伏尔泰(Voltaire),都认为人的大脑足以理解人类社会的工作方式。这导致了哈耶克所谓的铸成大错,如法国和布尔什维克的革命。其时,自上而下的政治权力以公正社会的预设重整社会。在哈耶克的时代(20 世纪的中期),这个错误不仅发生在社会主义国家,如依赖理性计划和中央集权的苏联,还发生在欧洲的社会民主党执政的福利国家。
在哈耶克看来,错误原因很多,最重要的是没有一名计划者,能掌握足够的社会实际运作知识,以作出理性的重新安排。社会中的知识,大部分具有本地特性,再向整个社会扩散,没人能掌握足够信息来预测法律或规则改革后的效果。⑬
哈耶克认为,社会秩序不是自上而下的理性计划的结果,而是在数百或数千分散个人的互动中自发产生的。那些个人尝试各式规则,保留有效的,拒绝无效的。社会秩序产生的过程是递增、进化、分散的,只有借用无数个人的本地知识,有效的“大型社会”方能出现。自发的秩序获得发展,以达尔文为生物有机体所安排的方式——分散的适应和选择,并不倚靠创世主的专门设计。
在哈耶克看来,法律本身便构成一种自发秩序,“毫无疑问,人们发现可以制作或更改法律之前,它已存在良久”。事实上,“个人学会观察(和遵守)行为规则之后的很久,才用语言将之表述出来”。立法——有意识颁布的新规则——“发生于人类历史的相对晚期……所有的法律都是、能够是、也应该是,立法者的自由发明……事实上,这是一种谬误,一个建构论唯理主义的谬种”。⑭
哈耶克心目中的自发秩序模型就是英国的普通法,无数法官设法将普遍规则用于所面对的特定案例,其判决的累积促使法律的进化发展:
英国人享有的自由在 18 世纪令其他欧洲国家的人们羡慕不已……它是这样一个事实的结果,即支配法院审判的法律乃是普通法。该法律独立于任何个人意志,它既约束独立的法院,又为这些法院所发展。对于普通法,议会很少加以干预,即便有所干预,其目的也主要是为了澄清某一法律系统内的疑点。⑮
哈耶克由此锁定法治的本质:代表整个共同体愿望的既存法律,高于当前政府的意志,它限制着政府的立法范围。他对英国普通法的偏爱,获得当代经济学家的赞同,他们也认为,它比欧洲大陆的民法传统更为灵活,对市场更为友好。⑯
哈耶克在解说其法律起源理论时作出两项声明,一项是实证性的,另一项是规范性的。他主张在大多数社会中,法律以自发的进化方式发展,这种自然生成的法律应该优于有意识制定的法律。这一解释也是伟大的英国法学家爱德华·柯克(Sir Edward Coke)所推崇的,他认为普通法始于太古时代。埃德蒙·伯克(Edmund Burke)在为渐进主义(Incrementalism)辩护时,也援引此一解释。⑰哈耶克是强大国家的伟大敌人,不管是苏联风格的共产党专政,还是以再分配和调节来实现“社会公正”的欧洲社会民主政体。在法律学者罗伯特·埃里克森(Robert Ellickson)所谓的“法律中心论”和“法律外围论”的长久争论上,哈耶克立场鲜明地站在后者一边。前者认为,正式制定的法律创立和塑造了道德规则;后者主张,它们只是编纂了非正式的既存规范。⑱
然而,哈耶克对最低限度国家的规范性偏爱,扭曲了他对法律起源的实证性见解。在很多社会,法律的存在确实早于立法,但政治当局经常介入以作修改,甚至在早期社会也是这样。现代法治的出现全靠强大中央国家的执法,其显而易见之处甚至可在他偏爱的普通法的起源中找到。
从惯例法到普通法
哈耶克认为,法律在社会规则分散演变的基础上获得发展,这一基本见解在广义上是正确的,无论是古代还是现代。但法律发展有重要中断,只能以政治权力的干预来解释,而不是“自发秩序”进程的结果。哈耶克只是把历史事实搞错了。⑲
这些过渡中有一个是英国从惯例法到普通法的过渡。普通法不只是惯例法的正规文本,它们之间有根本的差别。如我们在第 4 章中看到的,社会从部落组织过渡到国家组织,法律的意义便发生了重大变化。在部落社会中,个人之间的正义有点像当代国际关系,以竞争团体的自助为基础,没有更高级别的第三方执法。相比之下,国家层次的社会恰恰有如此的执法者,那就是国家本身。⑳
罗马帝国终结后的英格兰仍是部落组织,由盎格鲁人(Angles)、西萨克逊人(West Saxons)、朱特人(Jutes)、凯尔特人(Celts)等组成,尚无国家。家庭组成村庄,村庄再组成所谓的百户(足以承受百户居民)或县的更大单位。该层次之上就是国王,但早期君主没有武力的垄断,也不能对部落单位执行强制规定。他们不把自己当作领土的统治者,只是民众的国王——如盎格鲁人的国王(Rex Anglorum)。如我们在上一章看到的,6 世纪末,本笃派(Benedictine)的修道士奥古斯丁(Augustine)抵达英格兰,基督教便开始破坏盎格鲁—萨克逊的部落组织。但部落法律受到的侵蚀只是日渐月染的,到公元第一个千年后半期的混乱时代,仍然盛行。亲戚团体内有深深的信任,但竞争氏族之间却有敌意和警惕。所以,正义牵涉亲戚团体之间的相处规则。
盎格鲁—萨克逊编纂的第一本部落法律,是公元 600 年左右的《埃塞尔伯特法典》(Laws of Ethelbert),与稍早的墨洛温(Merovingian)国王克洛维一世的《萨利克法典》非常相似,罗列出各种受伤的赔偿金:
四个前门牙,每个价值六先令;其旁边的牙齿价值四先令,剩下的牙齿价值一先令;大拇指、大拇指指甲、食指、中指、戒指手指、小手指,各自的指甲都有区分,分别定价。相似分类也用在耳朵上,损失听力、耳朵削掉、耳朵穿孔、耳朵割裂;用在骨头上,骨头暴裸、骨头损坏、骨头断裂、头骨打破、肩膀失灵、下巴断裂、领子骨断裂、手臂断裂、大腿骨断裂、肋骨断裂;用在瘀伤上,衣服外的瘀伤、衣服内的瘀伤、没显黑色的瘀伤。㉑
基于赔偿金的惩罚,其特征是不公平,因受伤者社会地位的不同而有差异。所以,杀死一名自由人的赔偿金,也许是杀死仆人或奴隶的好几倍。
日耳曼的部落法律在本质上类似于其他部落社会,从努尔人,到当代的巴布亚新几内亚一语部落。如果有人伤害了你或你的亲戚,你的氏族为了保护自己的荣誉和可靠性,必须施以报复。受伤和报复都是集体的,报复对象不一定是行凶者,他的近亲通常就也足够。赔偿金的存在就是为了解决争端,以防升级,成为无休止的血亲复仇或部落间的仇杀。
现代法庭的遥远起源就是调停血亲复仇的氏族聚会。在盎格鲁—萨克逊部落,这就是模拟法庭,倾听控告和被告的作证,然后商讨适当的赔偿。但它没有现代的传讯权利,以逼迫证人出庭。它的裁决也得不到执行,除非达成协议。法律的证据往往有赖于用刑,譬如迫使被告赤脚走过火红的煤炭或犁头,或干脆将他们扔进冷水和热水,看是沉还是浮。㉒
如尼采所观察到的,基督教传入日耳曼部落后,给道德带来了深远启示。基督教的英雄是和平圣徒和烈士,不是武士或报仇的征服者;其传道的普遍平等,又相悖于部落社会基于荣誉的等级制度。基督教有关婚姻和遗产的新规则,不仅破坏部落团结,还创造新社区观念,其成员不再忠于亲戚团体,而分享共同信仰。国王的概念也从共同祖先团体的领袖,变成广大基督徒社区的领袖和保护人。不过,这个改变是循序渐进的。
部落制在基督教社会中的消亡并不意味家族制的死亡。在东正教中,这段时期的主教和教士可以结婚生子,还可实行宗教名义下的纳妾(nicolaism)。教会通过信徒的捐献获取愈来愈多的财产。教会领袖争取将圣俸传给孩子,加入地方的氏族和部落的政治运作,都变得不可避免。教会职位经手这么多的财富,本身也变成可供交易的珍贵财产,该做法叫作圣职买卖(simony)。
日耳曼异教徒皈依基督教,就像阿拉伯或突厥部落社会中不信者之皈依伊斯兰教,向哈耶克自发秩序的理论提出了有趣挑战。浏览哈耶克的相关阐述,找不到点滴的宗教因素。然而众所周知,在犹太教、基督教、印度教、穆斯林的社会中,宗教是法律规则的重要来源。基督教进入欧洲,给刚从部落习俗中脱颖而出的惯例法带来第一次主要中断。婚姻和产权规则发生变化,允许女子拥有财产,但这不是地方法官或社区的自发试验,而是强大的天主教等级制度所颁布的革新。教会并不反映地方上不同的价值观念,东正教和穆斯林的宗教当局,都没以相似方式来改造社会上现存的亲戚规则。教会很清楚,它不只是在批准惯例法:教皇乌尔班二世(Urban Ⅱ)在 1092 年告诉佛兰德伯爵(Count of Flanders):“你宣称,你只不过是照地方上的古代习俗行事?即使如此,你应该知道,创世主说过:我的名字是真理;他没有说过:我的名字是习俗。”㉓
英国法律发展中第二个主要中断是普通法的引入。普通法并不是惯例法的自发演变,它与早期国家的兴起密切相关,并凭借国家权力而取得最终的统治地位。事实上在诺曼征服之后,向全国颁布统一的普通法,已变成扩展国家权力的主要工具。伟大的法律学者弗雷德里克·梅特兰和弗雷德里克·波洛克,如此解说普通法的起源:
国王法庭(royal court)的习俗就是英国的习俗,从而变成了普通法。对于地方习俗,国王法官以一般性的语言表示尊敬,我们并没有看到任何移风易俗的主观愿望。不管如何,地方习俗即使没遭破坏,也得不到成长。尤其是程序,国王法庭取得了对所有其他法庭的彻底控制,将自己的规则视为唯一公正的。㉔
弄不清早期欧洲国王的作用,就不能理解这个过程。11 世纪的国王不是领土统治者,更像分散封建秩序中伙伴中的老大。像威廉一世和亨利一世那样的国王,花大部时间在旅途中察看国土的各部分。其时,大家都已退回各自分隔的村庄和庄园层次的小社会,这也是国王宣告权力和保持联系的唯一方法。国王的主要服务是充作上诉法庭,若有人不满意领主法庭(seigneurial)或庄园法庭(manor)所提供的正义。从自身利益出发,国王也希望扩充自己法庭的司法权,因为它的服务是收费的。向国王法庭提出上诉增强国王的威望,他可以推翻地方领主的裁决,从而削弱后者的权威。㉕
起初,各类法庭相互竞争,以取得司法生意。随着时间的推移,国王法庭开始占据优势。人们避开地方法庭有多种原因。巡回的国王法庭被视为更加公平,与领主法庭相比,它与本地诉讼人的牵连更少。它们也有程序上的优势,如强迫民众参与陪审团的工作。㉖长年累月,它们又获益于规模和范围上的经济效益,司法需要人力、专长、教育。第一个全国官僚机构是国王法庭所建立的,它开始编纂惯例规则,建立先例系统。显而易见的,写作是必要的前提。每过十年,熟悉先例的法律专家越来越多,再被指定为法官,派往全国。
顾名思义,普通法就是不特殊,普遍适用。也就是说,英国不同地区的众多惯例规则,现由单一的普通法所取代。各地的先例适用于全国,即遵循先例的原则(stare decisis)。执法的是法官网络,其工作环境是统一的法律系统,比以前拼凑的惯例规则更为系统、更为正式。普通法基于惯例法所订下的先例,但国家权力的兴起,创造了惯例规则不敷使用的全新环境。例如,以前亲戚团体以赔偿金解决的犯罪,现受到更高级别的第三方的起诉,或是庄园主,或是国王本人。国王法庭也开始变成无争议事项的登记场所,如财产注册和土地转移。㉗
因此,普通法代表了英国法律发展的中断。它依据较早的先例,如果没有诺曼征服,绝不可能成为全国法律。诺曼征服赶走了古老的丹麦和盎格鲁—萨克逊人的贵族,建立起愈益强大的大一统中央政权。以后的普通法演变可能是自发的,但它作为法律裁决的架构,又需要中央政治权力的干涉。㉘
历史学家约瑟夫·斯特雷耶认为,中世纪时,早期国家的创建涉及法律制度和财政制度,而不是军事组织;军事动员促进国家建设,则要等到早期现代。在某种意义上,法律机构甚至早于财政机构,因为国王法庭是国王收入的最重要来源之一。国王提供平等正义的能力——不像惯例法中,依据受害者社会地位而定不同的赔偿金——加强了自己的威望和权威。㉙像中东传统中的君主,国王不一定被视为最大最具掠夺性的军阀。他又可充任受地方领主掠夺的牺牲者的保护人,一个主持正义的人。
中央国家的法律功能,对英国后来的产权发展和国家的合法性至关重要。对地方领主与自由佃户和非自由佃户的交易,领主法庭享有专门司法权,直到大约 1400 年。这种情形下,一旦发生财产争执,就有点像由狐狸来守护鸡笼。逐渐地,国王法庭宣称有权过问这些纠纷。13 世纪早期,有人提出国王在全国范围享有司法权,低级法庭的司法权来自国王的委托。原告偏爱把诉讼送到国王法庭,久而久之,领主法庭慢慢失去对土地租佃纠纷的司法权。㉚这一市场驱动的选择显示,国王法庭肯定被视作更加公平,更少偏向地方领主,更可能执行裁决。
其他欧洲国家没有发生类似的改变。尤其在法国,领主法庭保留对土地租佃纠纷的司法权,直到法国大革命。在某种意义上,这很讽刺。一般认为,17 世纪的法国国王,如路易十三和路易十四,明显不同于英国国王,通过坚持自己的绝对权力来削弱贵族阶层,但他们却把地方法庭的司法权留给省城贵族。亨利·梅因爵士在他的论文《法国和英国》中指出,革命爆发之后,全法国的庄园主住宅被烧,纵火的第一对象是储存财产文件的契约房(muniment room)。不像英国农民,法国农民觉得地主手中的地契不合法,由于地方领主控制的法庭一直抱有偏见。㉛
最后的案例点明了法治性质的要点。法治依靠法律本身和可见的管理机构——法官、律师、法庭等,也依靠制度运作的正式程序。但法治的正常运作,既是制度或程序上的事务,也是规范性的事务。和平社会中的大多数人服从法律,不是因为做了理性的利弊计算,恐惧处罚;而是因为相信法律基本上是公平的,在道德观念上已习惯于遵守。如果相信它是不公平的,他们就比较不愿服从。㉜
被视作公平的法律,如果执行不均,或有钱有势者得以豁免,也将被认作不公平。这似乎将负担重又放回制度和程序,以及其公平执法的能力。这里仍有规范化的问题,如果有钱有势者在某种程度上不相信自我约束的必要,甚至不相信有约束同类的必要,光是制度何以遏制他们?在很多法治软弱的国家,法官、检察官、警察可被收买,或可被胁迫,正式制度的存在又能发生什么效用呢?
要建立规范化的法律秩序,不但国王接受,老百姓也愿接受,宗教就很有必要。波洛克和梅特兰写道,国王并不在法律之上:“每个国家一定要有某人或某些人在法律之上,一名既无义务又无权利的‘君主’,这样的理论一定会遭到拒绝……没人假设,国王可以更改天主教会的普通法,即使获得高级教士和男爵的同意。”㉝国王受到约束,因为百姓会以造反来反对他们所认定的不公。什么是不公,什么会动员百姓起来反抗国王,全看国王的做法合不合法。㉞
即使是公平的规范化秩序,也需要权力。如果国王不情愿执行针对精英的法律,或心有余而力不足,法律的合法性就会受损,不管其来源是宗教、传统还是习俗,这是哈耶克和他的自由至上主义追随者所疏忽的。普通法可能是分散各地法官的业绩,倘若没有强大的中央国家,它首先不会形成,之后也得不到执行。 英国很早就完成了从惯例法到现代法律制度的过渡,让人印象深刻,这构成了国家合法性的基础。其他欧洲国家在 13 世纪完成类似过渡,但依据的是完全不同的法律制度,即来自《查士丁尼法典》的民法。欧洲大陆的过渡,其关键也是天主教会的行为。这个故事,以及教会如何不同于印度和穆斯林世界的宗教机构,将是下一章的主题。
第 18 章 教会变为国家
天主教会对法治在欧洲的形成至关重要;叙任权斗争和后果;教会获得国家般的特征;世俗统治领域的出现;当代法治植根于上述发展
最深刻意义上的法治意味着:社会产生共识,其法律是公正和既存的,能够约束其时统治者的行为;享有主权的不是统治者,而是法律;统治者的正当权力只能来自法律,方才享有合法性。
在我们的世俗现代之前,在政治秩序之外,公正法律的最显著来源是宗教。宗教权威只有独立于政治权威,基于宗教的法律才能约束统治者;如果宗教权威组织涣散,或国家控制着教会的财产及教士的任免,那么宗教法律更有可能是在支持而不是限制政治权威。要理解法治的发展,不但要看宗教规则的来源和性质,还要关注宗教权威的组成和建制化。
欧洲的法治植根于基督教。欧洲国家出现之前,罗马就有颁布权威法律的基督教主教(pontiff)。欧洲关于婚姻和遗产的规则,最初不是君主所规定的,而是来自像教皇格里高利那样的个别人士。他的特使奥古斯丁带着他一清二楚的指示,远赴不列颠岛,以说服异教的埃塞尔伯特国王皈依基督教。
激进伊斯兰主义在 20 世纪晚期兴起以来,很多人指出,西方的教会和国家截然分开,但是,像沙特阿拉伯那样的穆斯林国家却政教不分。但这一差别经不起仔细的推敲。自基督教出现以来,西方的政教分离并不是常数,而是时断时续的。
基督教起初只是一个千禧宗派,在其存在的头三个世纪受到残酷的迫害,先是犹太人,再是罗马政治当局。到公元 313 年,君士坦丁(Constantine)皈依基督教,它从非正统宗派一下子变成罗马帝国的国教。罗马帝国的西部遭到异教野蛮人的征服,宗教和政治权力又一次分开。西方政权的孱弱给予天主教更多的独立机会,教皇哲拉修一世(Gelasius,492—496 年在位)在教条中争辩,高级教士拥有比君主行政权更高的立法权。①到了黑暗时代之末,政治权力重新恢复,政教第二次交融。
政教合一(Caesaropapism)是一种制度,它的宗教权威完全服从于国家,像基督教向罗马国教的转化。现保留给教皇的最高教士(pontifex maximus)头衔,曾是罗马皇帝的,因为他也是罗马国教的首脑。中国始终是政教合一(唐朝可能是例外。其时,佛教在精英中颇受欢迎),此外还有什叶派掌控地区之外的大部分穆斯林世界。拜占庭的东罗马帝国是现代东正教的老祖宗,也是政教合一称号的发源地。它始终不变,直到土耳其在 1453 年征服君士坦丁堡。大家所忽视的是,到了 11 世纪初,西方基督教世界的大部都已变成实际上的政教合一。
政教合一的实际意义是指政治当局对教会享有委任权,中世纪早期的欧洲都是如此。全欧洲的皇帝、国王、封建领主都在任命主教,也有权力召开教会会议,颁布教会法律。教皇将合法性赋予皇帝,皇帝却也在指定和罢黜教皇。1059 年之前的二十五位教皇中,皇帝任命了二十一位,罢黜了五位。教会当局对文官当局的惩罚,欧洲国王都享有否决权。②
在多数欧洲国家中,教会确实拥有四分之一到三分之一的土地,从而得到收入和自治。由于政治当局控制了教会圣职的任命,教会的独立程度还相当有限。教会的土地经常被认为是皇家的赞助,统治者经常委任亲戚为主教,主教和教士又允许结婚,经常会卷入他们所管辖地域的家庭和宫廷的政治。教会土地可变成遗产,传给主教的孩子。教会官员也担任政治职位,进一步增强了宗教和政权的牵连。③所以,教会本身就是前现代的家族组织。
天主教会宣告独立
11 世纪晚期,天主教开始独立于政治权力。领衔带头的是一位名叫希尔德布兰德的修道士(Hilderbrand),后来他成为教皇格里高利七世(1073—1085 年在位)。④他在教皇派中凝聚了一帮人,包括彼得·达米安(Peter Damiani)、红衣主教汉伯特(Humbert)、教皇帕斯卡尔二世(Pascal Ⅱ)。他们认为,教皇应对所有的基督徒和政治当局行使至高无上的法律权力,并有罢免皇帝的权利。他还宣称,任命主教的唯一机构是教会,而不是世俗当局。其时背景是神圣罗马皇帝亨利三世(Henry Ⅲ)的阴谋诡计。为了出席加冕典礼,他抵达罗马,马上罢免作为对手的三位教皇,以推举自己的候选人。⑤
根据希尔德布兰德,教会一定要实施改革,才能独立于政治权力,最重要的一点就是要严禁教士、主教结婚和生儿育女。他攻击常见的宗教纳妾和圣职买卖,它们让教会职位变成了可供交易和遗传的财产。⑥希尔德布兰德派发起了一场传单战役,敦促基督徒不要接受已婚或纳妾教士的圣礼,并抨击为赚钱而提供教会服务的行径。⑦成为格里高利七世后,他把教士独身订为教会的正式原则,并迫使已婚教士在教会义务和家庭义务之间作出选择。这是向教士既得利益的挑战,导致教会内部艰巨而激烈的斗争。教皇格里高利的目标是想在教会内终止腐败和寻租,所以攻击家族制的根源,即主教和神父的生儿育女。他的思维逻辑无异于中国和拜占庭依赖太监、奥斯曼帝国从家人手中夺走军事奴隶。如果在忠于国家和忠于家庭之间作出选择,大多数人出于生物本能会选后者。所以,减少腐败的最直接方法,就是禁止官员组织家庭。
这项改革自然遭到现有主教的反对。教皇格里高利明白,他赢不了这场战役,除非他有权任免主教,而不是皇帝。在 1075 年的教皇宣言中,他要将罢免主教和世俗教职的权利从国王手中收回。神圣罗马皇帝亨利四世的答复,是要将他罢黜,“下台,下台,你这个受诅咒的”。格里高利的回应是将皇帝逐出教会。⑧很多日耳曼君主和一部分主教支持教皇,迫使亨利四世在 1077 年赶来格里高利在卡诺莎的住所。他足足等了三天,赤脚站在雪地,以求教皇的宽赦。
有些历史性事件全由个人引起,如不提及他们特殊的道德品质,就难以解释。叙任权斗争就是这样的事件。格里高利有不屈不挠的坚强意志,在教皇派中,曾被伙伴称作“我神圣的撒旦”。就像四个世纪后的马丁·路德,他对改革之后的教会以及其在社会中发挥的作用,抱有恢宏的远见。他不怕胁迫,愿意看到与皇帝的冲突逐步升级,直至全面对抗。
但这历史上的著名冲突,仅靠个人意志是解释不清的。天主教会成为自治的政治参与者,其重要背景是欧洲普遍的政治软弱。拜占庭的东正教及其在俄国的正统继承者,不得不接受其所在帝国的监护。相比之下,西方教会位于政治上分崩离析的意大利半岛,北方邻国的日耳曼人也是散沙一般,神圣罗马帝国只取得名义上的统一。11 世纪的法国并不团结,无法果断地干涉教皇政治。这段时期的教会虽然没有自己的军事力量,但很容易在周边政治体的相互竞争中合纵连横。
亨利四世在卡诺莎接受教皇的权威,但仍不愿承认教皇委任主教的权利,依旧拒绝格里高利的要求。他继续占领罗马,罢免格里高利,让自己提供的候选人克雷芒三世(Clement Ⅲ)成为一位对立教皇(antipope)。格里高利向意大利南部的诺曼国王们求救。他们答应,但到最后洗劫罗马,引起罗马居民的反抗。格里高利被迫与诺曼同盟一起撤回南方,于 1085 年死于萨莱诺(Salerno),身名俱败。叙任权斗争延续到下一代,格里高利的继承者,再将亨利四世和其儿子亨利五世逐出教会。另一方面,皇帝罢免教皇,扶持自己的候选人成为对立教皇。最终达成协议的是 1122 年的沃尔姆斯宗教协定(Concordat of Worms),皇帝基本上放弃叙任权,而教会承认皇帝在一系列世俗事务上的权力。
叙任权斗争对欧洲后续发展非常重要。首先,它允许天主教会进化成现代的、等级制的、官僚化的、依法而治的机构,如法律历史学家哈罗德·伯尔曼(Harold Berman)所认为的,还为后来建国者树立了榜样。根据亨廷顿,机构发展的标准之一就是自治,如果不能控制对自己官员的任命,机构就不可能是自治的。这也是叙任权斗争的中心争执。沃尔姆斯宗教协定之后,教皇变成教会等级制度中无可争辩的执行总裁,在红衣主教学院的建议下,可随意任免主教。
教会也纯洁自己的行止。教士的独身制消除了将圣俸授予亲戚和后裔的诱惑,并给教职出售涂上新的道德色彩。教会可以什一税(tithe)的形式征收税赋,由于教职人士脱离地方氏族的政治,而变得更善于处置自己的财政资源。它还显示出真正国家的特征,有时组织自己的军队,在确定领土中(尽管很小)行使直接司法权。
教会对世俗事务的介入,当然未因叙任权斗争而告结束。世俗统治者也在继续设法操纵教皇职位,安置自己的候选人,例如 14 世纪的阿维农教皇(Avignon)。随着时间的推移,又出现新式滥权,最终为宗教改革铺平道路。与世界任何其他宗教机构相比,天主教在适应性、复杂性、自治性、连贯性方面的建制化更为高级。
叙任权斗争的第二个重要成果是精神领域和尘世领域的明确分离,从而为现代世俗国家铺平道路。如早先提及的,这个分离只在基督教中隐性存在。沃尔姆斯宗教协定,在西方教会的历史上永远终止了政教合一时代。这种方式,从没出现于东正教或穆斯林世界。
为了削弱政治统治者的权力,格里高利的改革宣告教会的普遍权威,不管是精神还是尘世,甚至还包括罢免国王和皇帝的权利。事实上,基督教皇是在要求印度婆罗门从一开始就在行使的权威。然而,经过漫长的政治和军事的博弈,教会被迫妥协。它划出明确界定的精神领域,让自己实施无可争辩的控制,同时又承认,世俗统治者有权在另外范围行使统治权。这一分工,为后来世俗国家的兴起打下基础。⑨
最后,叙任权斗争对欧洲法律和法治的发展产生了重大影响。第一,教会阐述系统性的教会法规取得合法化;第二,教会创造了建制化的精神权威的单独领域。
罗马法的再现
与皇帝发生冲突时,格里高利和继承者没有自己的军队可以调动,只能通过呼吁合法性来加强自己的力量。于是,教皇派发动了一次对法律源头的搜索,以支持教会享有普遍司法权的主张。搜索结果之一是 11 世纪末,在意大利北部的图书馆内重新发现《查士丁尼法典》(Corpus Iuris Civilis)。⑩迄今,《查士丁尼法典》仍是民法传统的基础,不管是欧洲大陆,还是受其殖民或影响的其他国家,包括从阿根廷到日本。很多基本的法律常识,如民法和刑法、公法和私法之间的差别,都可从中找到起源。
《查士丁尼法典》是罗马法律高度精细的汇集,6 世纪初,在查士丁尼皇帝治下的君士坦丁堡成书问世。⑪重被发现的文本包含四部分:摘要、制度、法典、案例,其中摘要最为重要,涵盖的题目包括个人地位、民事侵权、不公平致富、合同、补偿。查士丁尼时代的法学家相信,它是早期罗马法(现已遗失)最重要遗产的汇总,并变成 12 世纪新一代欧洲法学家的研究主题。⑫
罗马法的复兴之所以可行,是因为在新式机构中开展了法律研究,那就是新兴的现代大学。11 世纪末,博洛尼亚(Bologna)大学成为研究中心,来自欧洲各地的数千学生聚集起来,聆听像伊尔纳留斯(Irnerius)那样的教授讲解摘要。⑬新的法律课程让欧洲人看到一套详尽的法律系统,可立即用于自己的社会。《查士丁尼法典》的知识由此传播到欧洲大陆最遥远的角落,法律学院在其他城市纷纷涌现,如巴黎、牛津、海德堡(Heidelberg)、克拉科夫(Cracow)、哥本哈根。⑭有点像英国普通法的情形,罗马法的恢复突然取代了盛行于欧洲的日耳曼惯例法,代之以更为统一的跨国规则。⑮
推介《查士丁尼法典》的第一代学者被称为训诂者(glossators),其主要工作是重建罗马法。后续一代的学者,如托马斯·阿奎那(Thomas Aquinas),则看得更远,为寻求法律的思想基础而直抵古希腊。亚里士多德等古典哲学家认为,习俗和见解需要接受人们的理性考量,并对照于更普遍的真理标准。阿奎那将这条原则,用于自己对亚里士多德的研究。他所建立的哲学传统,鼓励后代法律评论家不要机械复制现存法律,而要推论法律来源,以做到活学活用。⑯欧洲大学所复原的古典传统,不仅是向静态的文本寻求权威,而是对文本的涵义进行理性查询。
新兴大学培养了一批特别律师,既能解释古典文本,又掌握专门知识。教会和世俗的当局开始认为,他们需要依赖律师的专长来作出裁决,尤其是在极为重要的商业合同和产权方面。律师依次发展自己的机构利益,拒绝非专家和自私的政治派别闯入他们的专业领域。
格里高利改革之前,教会法律包括宗教会议的法令、教父的著作、教皇法令、代表教会的国王和皇帝所颁布的法令。此外,还混杂有罗马法的残余和日耳曼的惯例法。⑰随着教会等级制度的建立,教会第一次有可能权威地制定法律,凭借愈益专业的教会法律专家,将统一性注入新法典。受过法律训练的修道士格拉提安(Gratian),分析、校对、调和了数世纪以来的几千条正典(canons),再将之综合成统一的法规。这本《教会法规汇编》(Concordance of Discordant Canons)出版于 1140 年,洋洋洒洒一千四百页。格拉提安建立了神圣法、自然法、制定法、惯例法的法律等级制度,又设计了理性程序,以解决相互之间的矛盾。格拉提安之后的一个世纪,教会法规得到极大扩充,涵盖了广泛的法律题目,包括刑法、家庭、财产、合同、遗嘱。⑱
天主教会通过统一教会法规的概念而取得国家属性,又通过发展行政官僚机构,而变得更像一个国家。法律学者认为,韦伯所定义的现代官僚的“职位”(office),其第一个模型是在 12 世纪教会等级制度中产生的。⑲现代职位的特征之一是职位和官员的分离,职位不是私人财产,执掌职位的只是领薪官员,身受所处等级制度的纪律约束;职位依功能而分,执掌职位要有技术专长。如我们所知,所有这些都是秦朝以来中国官僚制的特征,尽管有不少“职位”在后续朝代中重新家族化了。教会的叙任权从世俗政权的手中获得解放,教士独身制又得到强行的实施,自此以后,教会的官僚制特征愈益明显。例如,教会开始在 12 世纪早期区分教职(officium)与圣俸(beneficium)。教职人士不一定收到封建圣俸,现只是领薪的教会员工,根据自己的工作表现或被雇用,或被辞退。这些官僚开始任职于教皇秘书处(Papal Chancery),很快又变成世俗统治者秘书处的榜样。⑳
法律和现代国家的兴起
9 世纪卡洛林王朝崩溃之后,权力四下分散。到格里高利改革时期,欧洲的政治秩序见证了逆转的开头。权力流向一系列的地区领袖,当地方领主在 10 世纪末纷纷建造城堡时,又受到进一步的分割。庄园——基本上自给自足的生产和军事的单位,以领主的城堡和土地为中心——变成整个欧洲的统治来源。这个系统之上又出现几家王室,如以法兰西岛(Île de France)为中心的卡佩家族(Capetians)、征服英国和意大利南部的各式诺曼男爵。他们只是比对手拥有更多土地,遂变成新型领土国家的核心。
格里高利的改革不仅向领土国家提供了官僚和法律的榜样,并鼓励他们发展自己的建制。世俗统治者负责领土内的和平和秩序,并提供规则以促进新兴商业。这导致了独特法律领域的形成,分别与封建、庄园、城市、长途贸易有关。哈罗德·伯尔曼认为,法律形式的多样化激发了司法辖区之间的竞争和革新,从而促进自由在欧洲的发展。尤其重要的是独立城市的兴起,它的自由人口和对外贸的依赖,刺激了对商业法律的新型需求。㉑
教会在建制上趋向独立,更刺激了封建社会其他领域的集团组织。在 11 世纪,主教杰拉德·德·坎布雷(Gérard de Cambrai)和主教阿尔德贝隆·德·拉昂(Aldabéron de Laon)创立社会等级一分为三的原则:贵族、神职人士、平民——即打仗者、祈祷者、支持前两者的劳作者。这些功能组织与地域没有关系,其为三个代表阶层的形成打下意识形态的基础。统治者定期召集各代表阶层,以批准征税和讨论国家大事。如后续章节所显示的,欧洲国家今后发展的是负责制政府还是专制政府,将取决于这些阶层能否顶住中央君主的压力。㉒
欧洲国家建设的特征之一,是很早就非常依赖法律。法律在国家制度成长方面,既是动机,又是过程。专家习惯于认为,战争和暴力是欧洲政治发展的主要动力。这在早期现代肯定没错,其时,专制主义的兴起与军事动员的财政需求休戚相关。但在中世纪,国家获得合法性和权威,靠的是分配正义的能力,其早期机构多为执法部门。
最能体现这一点的,非英国莫属。21 世纪初,我们习惯把英国及其衍生品美国,当作盎格鲁—萨克逊经济自由主义的家园,把法国当作中央集权政府的诞生地。然而在 14 世纪之前,这正好恰恰相反。所有的欧洲政治体中,英国国家是最集中最强大的,其基础就是国王法庭,以及它向全国提供正义的职能。到 1200 年,它已拥有常设机构,配置以专业或半专业官员。它颁布法令规定,与土地权有关的案例,一定要得到国王法庭的命令方可成立。它还向全国征税。㉓中央权力的证据就在《末日书》(Domesday Book,即《土地调查清册》),它的编纂在诺曼征服后不久,核查了国内每一郡的居民。㉔
当时已有了英格兰国家身份的雏形。1215 年男爵们在兰尼米德(Runnymede)对抗国王约翰,强行施加《大宪章》(Magna Carta)。他们这样做,不是作为只想为自己争取豁免权的军阀。他们期待统一的中央政府,通过国王法庭来更好地保护自己的权利。在这一点上,他们把自己当作更大社区的代表。㉕相比之下,法国其时比较分散,各地区之间有重要的语言和文化上的差异,国王筹集税赋,只能在法兰西岛周围的自己领地。
中世纪教会为法治树立先例
天主教会在 12 世纪成为现代官僚机构,并颁布统一连贯的教会法规,但这离现代法治还很远。法治牢固的发达国家,向政府统治提供合法性的通常是书面宪法。但这套法律并不起源于宗教权威,事实上很多宪法规定,在牵涉宗教的道德问题上必须维持政治的中立。现代宪法的合法性来自某种民主的批准程序。这套法律可被看作扎根于永恒或普遍的原则之中,在亚伯拉罕·林肯看来,美国宪法就是一例。㉖但多数现代宪法对其合法性的最终来源都有点隐约其词。㉗从实用角度看,那些原则的解释仍然取决于政治上的争论。到最后,借民主取得合法性的行政和立法的机构,其权力仍然要受制于借民主取得合法性的宪法。后者取决于更严格的社会共识,如某种形式的超多数选举。在最近发展中,各国政府也要受制于跨国法律机构,如欧洲人权法庭(European Court of Human Rights)和国际战犯法庭(International Criminal Court)。不过,与国家层次的法庭相比,它们的合法基础比较暧昧。㉘包括以色列和印度的自由民主国家中,宗教法庭仍在家庭法上享有司法权。但这只是例外,宗教权威不得参与法律制度是普遍规则。
那么,为何要说基于宗教的法律为现代法治奠下了基石?
宗教权威的分开存在,使统治者倾向于承认,自己不是法律的最终来源。弗雷德里克·梅特兰坚信,没有一位英国国王认为自己高于法律。但这不适用于任何一位中国皇帝,因为没有一条法律是他们承认的,除非是自己的金口玉言。在这方面,像印度的拉贾和刹帝利、阿拉伯和土耳其的苏丹,基督教君主同意自己身处法律之下。
在每个实行以宗教为基础的法律的社会中,政治统治者都制定法律,试图侵入宗教法律的领域。在许多情况中这种侵入是必要的,因为有很多方面宗教法律不敷使用,但最危险的侵入是针对原则的。早期现代欧洲的重要政治斗争(将在后续章节中作详细说明)涉及崛起的君主,他们凭借新颖的主权原则,将自己置于等级制度的顶部,以取代上帝。这些国王像中国皇帝,声称自己可单独制作法律,不受既存法律、习俗和宗教的束缚。成功抵制这些声称,重申法律的至高无上,那就是现代法治兴起的故事。法律本身可能还不够,所以又从宗教传统那里获得圣洁、自治和连贯性,从而更易实行这种抵制。
法律体现有关正义规则的广泛社会共识,如果明白这一点,那么中世纪法治和现代法治之间的中断,与其说是实质性的,倒不如说是表面上的。这也是哈耶克所说的法律早于立法的涵义。在 12 世纪的宗教年代,或在同时期的穆斯林或印度世界,社会共识往往通过宗教表述出来。与今天相比,那时宗教在日常生活中发挥更为重要的作用。宗教法律不是从外空掉入社会的,一开始可能伴随暴力和征服而至,再与社会共同进化,渐渐演变成本土的道德规则。㉙当时,宗教和世俗的领域互不分离,阐明社会共识就不得不使用宗教语言。在宗教扮演较为局限角色的今天,无可避免地,必须通过其他途径来确定社会共识,譬如通过民主选举。无论用宗教语言还是世俗语言,法律始终是广泛分享的正义规则的表述。
12 世纪浮现的宗教法律,对现代法治施加了重大影响,它帮助促进了法律的建制化和理性化。法治若要存在,光是建立统治者服从法律的理论原则还不够。还要有体现有关法律的具体机构,并取得独立于国家之外的某种程度的自治,否则就很难控制国家的随心所欲。此外,如果法律不是一套连贯和清晰的规则,就不能限制行政权力。宪法上的分权,必须依靠一个切实的法律体系,该体系掌控自己的用人和晋升,设立自己的专业标准,训练自己的律师和法官,在解释法律时,享有不受行政机构干涉的真正权力。英国国王负责创建了以国王法庭为终极权威的普通法,他也将大量权力下放给法官,允许法律专业的茁壮成长,其就业和收入并不完全依赖国家。在欧洲大陆,查士丁尼的民法传统,意味着较为集中的法律诠释,但也有自治的法律专业的平行成长——事实上,出现了多种法律的多门专业。两种情形中,西方法律的理性化程度都要大于印度或穆斯林逊尼派。后两种传统文明中,没有涌现像修道士格拉提安那样的人,将整套既存的宗教法令统一连贯起来。
西欧出现的法律传统明显不同于东正教。影响后来政治发展的不是基督教本身,而是西方基督教所采用的特别制度。东正教的主教继续接受皇帝或本地统治者的任命,教会在总体上也从没宣告自己的独立。不像西方的教会,东正教从未丧失罗马法的传统,也从未宣称法律有高于拜占庭皇帝的至上地位。
法治的涌现是构成现代政治发展的三大组件中第二个。跟确定欧洲脱离部落或亲戚社会组织的过渡时间一样,法治出现的时间也需要再往前提,其远远早于早期现代时期——至少要提到到 12 世纪。这也点出了本卷的中心主题,即现代化的不同组件,并不全是某种一揽子解释的一部分,它们并非都是伴随宗教改革、启蒙运动和工业革命而来的。独立城市和新兴贸易的需求,促使了现代商业法律的发展。但法治一开始不是经济力量的产品,而是宗教产品。所以,作为经济现代化关键的两个基本制度——可以自由选择个人的社会关系和财产关系,透明预知的法律为政治统治设限——都是前现代中世纪教会所创造的。只是到了后来,这些制度证明在经济范围内也相当有用。
第 19 章 国家变为教会
法治在印度和中东的发展,但在中国缺席;中东世俗和宗教的当局有效分享权力;前现代中东政权遵守产权;穆斯林乌里玛不能以基督教会的方式制衡国家权力;当代阿拉伯世界没有法治;现代法治的比较
在中国,宗教并不反映社会和文化的共识,毋宁说是社会抗议的手段。这体现在汉朝的道教、唐朝的佛教、19 世纪受基督教影响的太平天国等。中国的国家轻易掌控各式祭司团体,从不承认比国家本身更高的宗教权威。
所以,中国没有基于宗教的法治的历史基础。中国的传统以法家思想为基石,中国人心目中的法律主要是制定法(positive law),也就是皇帝所颁布的王法。秦、汉、隋、唐、明等朝都出版了重要法典,很多篇幅只是各式违法的处罚表。7 至 8 世纪陆续颁布的《唐律》,不提法律的神圣来源,只说法律是世俗统治者所制作的,以控制百姓的行止和避免自然和社会的失衡。①
印度则完全不同,与印度国家形成同期或稍早的婆罗门教,规定政治/武士阶层——刹帝利——必须从属于祭司阶层的婆罗门。印度宗教以四大社会阶层的瓦尔纳为基础,印度统治者必须向身处顶端的祭司取得合法性和社会支持。所以,法律深深植根于宗教,而非政治。最早的法律文本《法论》(Dharmasastras),不是像中国那样的皇帝法令,而是宗教权威所写下的文本。②印度后来的法律发展有点像英国的普通法,没有严格遵循这些法律文本,反而依据判例,并把班智达(panditas,精通宗教典籍的学者)所创造的先例前后连接。③执行裁决的经常是婆罗门,而不是政治当局,不允许分开的世俗领域来制订规则。法律有很多哈耶克提及的特征,通常是不可更改的,除非能找到与当前法律有关的更古老先例。④独立后,印度议会试图修改婚姻和离婚的法律,据称有名保守印度人这样说:“议会的权力不可推翻经典(Shastras)的命令,那是上帝说的话,由圣人(Rishis)为我们抄录下的。印度人不可接受经典之外的任何权威。”⑤
然而,婆罗门阶层没有组织成单一的等级制度,不能对国王和皇帝发号施令,没有印度教皇,也没有印度教会。婆罗门阶层仅代表一个网络,其成员居住在无数的村庄和城市,彼此联络而已。婆罗门内部又分出不同的迦提,由此而充满等级差别。主持国王授权仪式的婆罗门,可能不愿与主持葬礼仪式的交往。宗教权威在地方上享有极大影响,几乎每一项社会事务都需要他们的服务。他们从不臣服于国家,或成为国家的雇员,但也无法凭借建制化的等级制度来采取集体行动。迦提所造成的权威碎片化,不单影响政治权力,也影响宗教权力。
中东的法治
除了印度和欧洲,出现法治的另一个世界文明是伊斯兰教的中东。今天,不管是境内还是境外的很多人都知道,那里的很多政权是残酷的独裁专制政府,尤其是在阿拉伯世界内,不受任何更高法律或正义的约束。⑥西方人通常认为,教会和国家的交融合一是伊斯兰教的本质,对基督教欧洲来说,才是天方夜谭。伊朗 1979 年革命后所建立的神权政府,只是返回传统的穆斯林统治。但这一切都不准确。
现代穆斯林独裁专制政府的出现是偶然事件的结果。这个偶然就是该地区与西方的碰撞对峙,以及之后向现代性的过渡。在基督教的欧洲,政治和宗教的权力经常联合起来。在穆斯林世界,它们在历史上很长一段时期倒是有效隔离的。法律在穆斯林世界中扮演的角色,与在基督教领土上的完全相同:制衡政治统治者的随心所欲——虽然较弱。法治是穆斯林文明的基础,实际上它在很多方面定义着这一文明。
让我们总结一下法律在穆斯林和基督教世界的社会作用的相同之处。在这两个传统中,法律都植根于宗教,只有一位上帝,行使普世的司法权,是所有真理和正义的源泉。这两个传统,再加上犹太教,都深深倚靠宗教的经典,其基本社会规则很早就被编纂成书。在伊斯兰教中,这些规则不仅是神圣的《古兰经》,还有圣行(sunna)和圣训(hadith),后者是穆罕默德生前的故事和训话,可作人们行为的指针。但这些规则的解释,在许多情况中又是模棱不定的,必须拜托专门的教士阶层——基督教中的牧师和伊斯兰教中的乌里玛(宗教学者)。在穆斯林和基督教世界,法律并不像中国那样出自政治权力,而是来自对政治当局享有统治权的上帝。穆罕默德生前可能已是部落的统治者,但在阿拉伯伙伴的眼中,他的权威并不在他所指挥的军队,而在他是上帝启示的使者。
跟穆罕默德一样,最初几位哈里发集宗教和政治的权力于一身,这在倭马亚朝代始终如此。该朝代结束时,政治和哈里发的权力才开始分隔。其时,倭马亚王子逃离阿拔斯王朝,在西班牙建立了分立的西方哈里发政权。阿拉伯帝国的不同省份,随着岁月的消逝而逐一分离出去,哈里发的权力只达首都巴格达和周边地区,甚至变成掌权军事指挥官的傀儡。⑦法蒂玛王朝(Fatimids)先后在突尼斯和埃及分别建立分立的哈里发政权。巴格达哈里发的权威从没获得什叶派和哈瓦利吉派的承认。哈里发可以宣称享有普遍的精神权威,但其真正的司法权非常有限。
到了 11 世纪,哈里发和在领土中行使政治权力的人分享权力。真正的掌权者——世俗君主——披上了“埃米尔中的埃米尔”的头衔。通过立法上的巧立名目,哈里发声称把世俗权力委托他人,以换取自己在狭窄宗教事务中的权威。⑧中世纪伊斯兰教法律学者艾布·哈桑·马沃尔迪(Abu al-Hasan al-Mawardi)解说这是合法的,因为哈里发通过代理人仍在行使世俗的权力,真相恰恰相反,哈里发只是埃米尔的傀儡。⑨伊斯兰教的世界实质上是政教合一,而不是神权。世俗统治者掌控权力,请哈里发和乌里玛来到自己领土,帮助管理伊斯兰教法。⑩
在逊尼派穆斯林世界中所缺乏的,恰好是哈里发和乌里玛脱离政治,发展成为分立的单独机构,享有分明的等级制度、司法权、人事权。也就是说,没能建成单独的穆斯林“教会”,可与格里高利改革之后涌现的天主教会媲美。跟叙任权斗争之前的天主教会一样,穆斯林知识阶层只是分散的网络,由教士、法官、阅读和应用穆斯林判例的学者所组成。逊尼派的传统内,有四家主要的穆斯林法律学派,相互竞争,在哲学上各持己见,其地位起伏有赖于权力的惠顾。乌里玛一直没有形成建制化的等级制度,无法建成单独法律传统和穆斯林等级制度,以罗马教皇的方式向政治权力提出挑战。
国家与清真寺的分离
但这并不意味宗教和世俗权力之间没有功能的分离。图森·贝(Tursun Bey)写道,15 世纪的奥斯曼帝国,苏丹可在伊斯兰教法之外自行制定世俗法律。这套世俗法律叫作卡奴纳莫(kanunname,该词源自欧洲使用的 canon law [教会法]),用于传统伊斯兰教法鞭长莫及的领域,如公共和行政的法律。所征服领土的征税和产权、发行货币、贸易管理,全靠这套世俗法律。⑪传统的伊斯兰教法主要涉及婚姻、家庭、遗产和其他私人事务,由教法专家卡迪和穆智泰希德(kadis and mujtahids)执行。他们熟谙穆斯林经典,能将这一庞杂的法典应用到特定案例,很像印度的班智达。⑫这就需要平行的两套司法建制,一个是世俗的,另一个是宗教的。卡迪应用伊斯兰教法,但其裁决必须依赖世俗当局的执法。⑬
在理论上,奥斯曼帝国日益增长的世俗法律从属于伊斯兰教法,需要接受宗教权威的审阅。哈里发在理论上高于苏丹,但在实际上却依赖苏丹。同样道理,因为日益增长的商业社会需要越来越多的规则,实际上的宗教法律反而遭受排挤。等到奥斯曼法庭设立大穆夫提(grand mufti,教法说明官)一职时,宗教权威的独立受到更大限制。以前,政府从学者圈中选任教法执行官卡迪,让他们自主处置法律内容。新的大穆夫提和他的属下,现在有权就伊斯兰教法的内容,发布不受限制的意见或论断(fatwas)。土耳其愈益增加对宗教的政治控制,所走的方向与欧洲恰恰相反。⑭如果说罗马教会展示出国家特征,土耳其国家则展示出教会特征。
前现代的中东究竟在什么程度上遵守法治?如第 17 章所提到的,今天普遍认可的法治至少有两层分开的意义:第一,遵守产权和合同的法律,允许商业和投资的发生;第二,统治者和统治阶级自愿接受法律所规定的限制。第二层意思直接影响第一层,如果社会精英不遵守法治,使用权力随意攫取弱势群体的财产,便成为巨大的诱惑。如前所述,统治者仍有可能在实践中遵守日常法治,但在理论上却有任意侵犯产权的权力。
对我们深入研究的两个中东政权来说,即埃及的马穆鲁克和土耳其的奥斯曼,第一意义中的法治作为预设条件而存在。也就是说,它们有关于财产和遗产的完善规则,允许长期的投资和可预知的商业交易。第二意义中的法治也同样存在,马穆鲁克和奥斯曼苏丹都承认,他们的权力受上帝创建的既存法律的限制。但在实践中,他们在解释法律以袒护自己私利时,仍享有相当大的余地,尤其在财政严峻时期。对税收的迫切需求,促使他们违反长期的法律规范。
但这两个案例都没有完全的现代产权,现代产权的付之阙如是否限制了穆斯林世界的经济发展,这不很清楚。⑮奥斯曼帝国拥有大量土地,分配给提供军事服务的骑士。替骑士耕种土地的农民,可把自己的使用权传给孩子。手艺人和商人等其他百姓享有私人产权,如果幸运和技术精湛,可积累大笔财富。所有传统的中东统治者,非常清楚苛捐杂税的危险,尽可能以“正义”名义予以回避。此外,他们像其他君主一样,把自己视作保护人,使平民免受贵族精英本能上的掠夺。甚至苏丹也不可越过法律。如果苏丹的骑士遵命来执行处罚,他们仍需要把被控者带到卡迪那里,以取得法律的裁决。如个人去世而未留遗嘱,财产在国家能够拿走之前必须由理论上的遗嘱执行者保管。非穆斯林的外国人过世后,其财产同样由法学家记录下来,直到继承人出现。⑯
法律如何限制传统穆斯林政府的权力,可在慈善性质的瓦克夫的作用中找到明显证据。如我们所知,掌权的奴隶军精英最初不可拥有后裔,也不可积累财产。马穆鲁克和土耳其禁卫军,首先避开规则以组织家庭,然后再设立慈善基金,安置自己孩子或亲信来运转这些基金,其收入将保证后代的生计。阿拉伯和土耳其的统治者,让这些瓦克夫完整无缺地持续数代,但有对改动遗产的严格限制,从而束缚了它们的经济效率。⑰
如果瓦克夫限定了国家攫取私人财产的能力,它的频繁使用意味着,其他不受宗教保护的财产往往面临随意的征税。尽管不是每个国家都堪称匪寇,但如有紧急情形,所有国家都可能成为掠夺者。15 世纪的切尔克斯系马穆鲁克政权,随着岁月的流逝,而陷入愈益可怕的财政困局,导致苏丹寻求火烧眉毛的计策以增加收入。他们任意提高税率,截获各种财富,导致富人寻找越来越具创意的方法来隐藏财产,不愿做任何投资。同样,奥斯曼在 16 世纪后半叶面临财政危机,导致税率增长,并威胁到传统产权。禁卫军职业的制度化老规矩,不得成家的禁令,都被一一放松。国家的封地不再留作军事服务的报酬,而被腐败当权人售给出价最高的投标者。像基督教统治者时时觊觎修道院的财富和其他教会财产,马穆鲁克甚至也突袭瓦克夫来筹措资金。
教皇的师团
据说,斯大林曾鄙视地问:“教皇手下有多少师团?”如我所说,既然法治植根于宗教,我们可向法官和律师提出一个类似的问题:他们在法治国家中部署了多少师团?他们凭什么来迫使统治者服从他们所解释的法律?
答案当然是零,行政部门和司法部门之间的分权只是隐喻性的。行政官拥有强制权力,可召集军队和警察来执行他(她)的意志。司法部门的权力,或身为法律监护人的宗教权威,体现在可向统治者提供合法性,以及作为社会共识保护人而获得广泛支持。格里高利七世可迫使亨利四世来卡诺莎,但实际上无法罢免这个皇帝。对此,他必须依赖军事同盟,比如嫉妒亨利四世的日耳曼君主和意大利南部的诺曼国王。教皇能否吸引世俗的同盟,则要依赖其事业的合法性,以及他们为自己短期利益所打的小算盘。叙任权斗争的结果是个复杂的混合体,既有物质因素,也有道德因素。最终,拥有军队和经济资源的世俗统治者,被迫与具有部分经济资源但全无强制权力的精神领袖达成妥协。教皇的权威确实存在,并不依赖他的师团。
穆斯林乌里玛的权威在于可向苏丹授予合法性,就像教皇的权威。遇上继承权的斗争,这种权威就变得非常重要。在穆斯林世界,伊斯兰教和突厥部落习俗,都反对建立王朝继承的明确规则,比如长子继承权。苏丹可指定继承人,但实际的继承过程经常变成一场苏丹儿子的自由参赛,或在马穆鲁克的情况中,变成一场主要派系领袖的自由参赛。在这种情境下,乌里玛给予或保留其支持的权力就是举足轻重的。如果权力斗争中的干预变得太公开,像切尔克斯系马穆鲁克时期的哈里发事件,他们可能会搬起石头砸自己的脚。
然而,我们不应夸大法治在前现代穆斯林社会中的作用。在保护产权和商业上,法律的运作尚属“足够好”,但提供不了像宪法保障的东西,以对抗存心违法乱纪的统治者。大穆夫提和卡迪都是国家选择和雇用的,明显减弱了他们的自治性,全然不同于 12 世纪之后天主教会聘请的独立法官。奥斯曼国家从头到尾都是政教合一,随着时间的推移,对穆斯林学者的控制程度日益增加。
印度和伊斯兰教的法治无法幸免于西方的叩门
在变成殖民地或接受西方重大影响之前,印度和中东的法治互相之间有很多类似之处。它们都有传统的书面法律,仰承宗教权威的保护,还有数世纪宗教法官(印度的班智达和穆斯林的卡迪)所积累的判例,作为先例而被继承下来。它们的宗教法律都是正义的最终来源。至少在理论上,政治统治者获得授权或代理权来执政。
印度和中东在这一方面,与基督教欧洲的距离,远远近于这三个地区与中国的距离。它们不同于欧洲的地方,在于其宗教机构都没有脱离政治秩序。婆罗门教中从来没有教皇,穆斯林的哈里发在倭马亚王朝之后,基本上成为伊斯兰地域中执政统治者的俘虏。这两种宗教机构不能独立于政府,也就无法发展成为自主控制用人和晋升的现代等级制官僚机构。没有自治,宗教法律的机构难以对国家发挥强大制衡。宗教机构与国家相互渗透,国家本身也不能发展成单独的世俗机构。
不管是印度还是穆斯林世界,传统的法治都没能在现代化之后继续幸存,对后者来说尤属悲剧。在 1772 年的印度,以瓦伦·哈斯丁斯(Warren Hastings)为首的东印度公司管辖区,决定将印度的法论用于印度教徒,将伊斯兰教法用于穆斯林,将英国版本的“正义、公平、良心”的法律用于其他案例。⑱在应用“印度教法”时,英国人误解了法律在印度社会中的作用。他们相信,法论(Dharmasastra)相当于欧洲的教会法,也就是,与世俗法律相对的、纂成法典并统一适用于所有印度教徒的宗教法。如我们所知,欧洲的教会法规发展至今,经历了漫长演变,但印度法律从没有过类似的进化。它与其说是基于文本的法律,倒不如说是一套鲜活衍变的规则,接受班智达的审视,依据语境而用于印度不同区域。⑲此外,英国统治者还因阅读梵语的能力有限而跌跌撞撞。英国人起初把班智达当作法论专家使用,随着更多梵语文本译成英语,遂改持不信任和回避的态度。班智达的使用到 1864 年完全废除,取而代之的是英国法官,全靠自己来设法解读传统的印度教法。(用于印度穆斯林的伊斯兰教法也遇上同样的中断。)⑳此时,作为活的传统的印度教法全然崩溃,到了印度共和国方才复兴,但传统的连续性已被腰斩。
穆斯林的法治传统发生更为彻底的中断。奥斯曼政府像英国人对待印度法律那样改革伊斯兰教法。它从 1869 年到 1876 年编纂了马雅拉法典(Mecelle,又译麦吉拉)。其目标是整顿伊斯兰教法,将之汇集成统一连贯的法典,以期达到 1140 年格拉提安整理基督教法规的效果(编按:参见本书第 18 章)。在这个过程中,他们削弱了乌里玛的传统社会作用。因为与灵活不定的体系相比,在严密编纂的体系中,法官作用完全不同,其重要性下跌。1877 年的奥斯曼宪法将伊斯兰教法降为各种法律之一,剥夺了它赋予政权合法性的作用。接受西方法律训练的法官,逐渐取代传统学者阶层。凯末尔(Kemal Ataturk)和土耳其共和国兴起于第一次世界大战之后,废除伊斯兰王朝,以世俗民族主义取代土耳其国家的伊斯兰基础。㉑阿拉伯人从不接受马雅拉法典的完全合法性,随着奥斯曼和青年土耳其党人等运动的展开,认同感的分裂日益增强。独立之后,他们发现自己陷于尴尬境地,一边是已简化的传统伊斯兰教法,另一边是殖民者带来的西方法律。
从殖民地走到独立之后,印度和阿拉伯的途径分道扬镳。印度共和国建立了宪法秩序,行政权力接受法律和立法选举的限制。独立后的印度法律一直都其貌不扬——像是现代和传统法律的拼凑物,以讲究程序和慢条斯理而声名狼藉。但它至少是一套法律,除了 20 世纪 70 年代英迪拉·甘地(Indira Gandhi)宣布的短暂紧急状态,印度领袖愿意在它的约束下运作。
阿拉伯世界走上截然不同的道路。英国、法国、意大利的殖民当局,其安插在埃及、利比亚、叙利亚、伊拉克的传统君主,很快被世俗的民族主义军官所取代。后者继而组织强大的中央政府,不受立法机关和法庭的限制。在这些政权当中,乌里玛的传统作用均遭废除,换成来自行政机构的“现代化”法律。唯一例外是沙特阿拉伯,它从没沦为殖民地,维持新原教旨主义(neofundamentalist)的政权,其行政权力受到瓦哈比派(Wahhabi)宗教机构的制衡。很多行政权力高于一切的阿拉伯政权,蜕化成压制性的独裁,无法为国民提供经济增长或人身自由。
法律学者挪亚·费尔德曼(Noah Feldman)认为,21 世纪早期的阿拉伯世界,伊斯兰教重新兴起,人们纷纷要求返回伊斯兰教法,既不满意当代威权政府的无法无天,又在怀念行政权力曾经尊崇法律的旧时代。他声称,回到伊斯兰教法的呼吁,与其说是反拨时钟,倒退回中世纪的伊斯兰教,倒不如说是在祈求政治权力遵守规则的平衡社会。反复诉求“正义”,甚至融入很多伊斯兰政党的名字。这不是在追求社会平等,而是在追求法律面前的人人平等。现代的强大国家,如果没有法治或负责制的制衡,能够成功实施完完全全的暴政。㉒
现代伊斯兰主义者能否建成接受法治制衡的民主政权?这是个很微妙的问题。1979 年革命后,伊朗伊斯兰教共和国的经验差强人意。自从 19 世纪以来,什叶派的伊朗一直拥有组织良好的神职等级制度,胜过逊尼派世界中任何其他组织。它在霍梅尼(Khomeini)阿亚图拉的领导下,夺取伊朗政权,建起真正的神权国家,政府部门都受神职人员的控制。该国发展成为神职的独裁政府,监禁和杀害政治对手,为达目的甘愿徇私枉法。
在理论上,伊朗共和国 1979 年宪法可以是温和、民主、守法国家的基础。它允许立法机关和总统的选举,但要接受限制。限制来自一名非民选的最高领袖,以及代表上帝的高级神职人员所组成的监督委员会(Guardian Council)。此类安排不一定是“中世纪”或前现代的。马克斯·韦伯认为是现代理性国家典型的德意志帝国(Wilhelmine Germany),其宪法规定要有民选的立法机关,但受非民选的恺撒的制衡。如果伊朗的最高领袖或监督委员会,把自己当作高级的传统乌里玛,享有类似最高法院的权威,不时宣布民选伊斯兰会议(Majlis)的立法不符伊斯兰教法,那么将之称作新式的伊斯兰教的法治,这还有一点道理。然而,1979 年宪法赋予最高领袖的,不仅是司法权,更是实质性的行政权。他控制伊斯兰教革命卫队军团和民兵(Basij),主动干涉让选举候选人丧失资格,操纵选举以制造有利结局。㉓像俾斯麦(Bismarck)宪法,或模拟它的日本明治宪法,伊朗宪法特地保留部分行政权力,不是给皇帝,而是给神职等级制度。与在日本和德国发生的情形一样,这种行政权力使人堕落,军队因此而加强对知识阶层的控制,恰恰与宪法所规定的相反。
国家建设旨在集权,法治却在一旁掣肘。因此,法治发展将遭遇政治竞争,并受制于特殊参与者的政治利益,如早期英王、雄心勃勃的教皇、要求回到伊斯兰教法的伊斯兰反对派。欧洲法治的基础始建于 12 世纪,其最终巩固还得有赖数世纪的政治斗争。后来,法治的故事开始与负责制政府兴起的故事水乳交融,因为负责制政府的倡导者不但要求民主选举,还要求行政部门遵守法律。我将在第 27 章再次讨论这个故事。
西欧的法治为何较强
过渡到现代化之前,法治便存在于中世纪的欧洲、中东、印度。这些社会的统治者承认,必须在并非由自己创造的法律下过活。然而,限制他们行为的实际程度,不仅取决于理论上的认可,还要依赖立法和执法的建制化状况。要想让法律对统治者构成更为有效的约束,需要某些特定的条件:它被编纂成权威的文本;法律的内容不由政治当局而由法律专家来确定;最后,法律被有别于政治等级的建制性秩序所保护,拥有自己的资源和任免权。
与中东或印度相比,西欧的法治获得更大程度的建制化。这与其说是宗教思想的缘故,倒不如说是欧洲发展中历史性的偶然情势所致,因为东正教就从未有过类似的发展。一个重要因素是欧洲权力的极端分裂,给了教会极大的机遇。这导致了颇不寻常的情形:法治得以在欧洲社会中生根发芽,不但早于民主和负责制政府的出现,而且早于现代国家的构建。这在建制化法律的方方面面都是昭然若揭的。
编纂
印度的“吠陀本集”口传心授,到后期方才写成文字。明显不同的是一神的犹太教、基督教、伊斯兰教,很早就开始以权威的经典为基础。他们都被称作“圣书上的民族”。但只有在西欧,混乱的文本、法令、解释和评论被梳理成逻辑统一的整体。在穆斯林、印度和东正教的传统中,找不到《查士丁尼法典》和格拉提安的《教会法规》的等同物。
法律专业化
在这一方面,基督教与其他传统基本上大同小异,大家都培养了解释和执行法律的专家。只是法律教育在先进大学系统中获得的开发和正规化,西欧要胜过其他地方。
机构自治
按照亨廷顿的分类,自治是机构发展的典型特征。在这一方面,跟其他地方相比,西方法律获得更多进展。世界其他地方都没有类似格里高利改革和叙任权斗争的经历。其时,整个基督教会机构都投入与世俗统治者的持久政治冲突,造成势均力敌的僵局。最后的沃尔姆斯宗教协定,确保教会作为一个机构的自治地位,并大大鼓励它发展自己的官僚机构和正式规则。
所以在前现代,与中东、印度和东正教相比,西欧的法治对世俗统治者的权力实施了更为强大的制衡。就后来自由制度的发展而言,这个意义重大。
欧洲的法治得以存活下来,尽管它的合法性基础在向现代化的过渡中发生了变化。这是内部有机发展的结果,宗教改革破坏了教会权威,启蒙运动的世俗思想又腐蚀了当时的宗教信念。基于国王、民族或人民的新主权思想,开始取代上帝的主权,而变成法律合法性的基础。许多评论家指出,西方法治比现代民主足足早了数个世纪,所以 18 世纪的普鲁士可以成为一个法治国家(Rechtsstaat),在人民主权原则获得承认之前,已在制衡行政权力。到 19 世纪的晚期,民主思想获得合法性,法律越来越被视为民主社会的正面措施。此时,法治所造成的习惯已在西方社会深入人心。文明生活与法律共存的观念、强大自治的法律机构的存在、资本主义繁荣经济的需求,合在一起加强了法治,尽管其合法性的基础已有变更。
我反复强调,一个没有法治的伟大世界文明是中国。中国皇帝当然有能力实施暴政,如秦始皇以法家的严刑峻法为基础创建大一统国家。然而,中国历代皇朝并不以严酷统治著称。在有关产权、征税及为重塑传统社会风俗而行干预的程度上,中国国家遵守明确的限制。如果这些限制不是来自法律,那源头到底是什么?作为成熟的农业社会,中国如何治理?这是下面两章的主题。
第 20 章 东方专制主义
唐朝之后,现代国家重获巩固;女皇帝武则天的篡位和从中透露出的中国政治制度;天命和政治合法性在王朝中国的确立
在王朝中国,没有皇帝承认法律权威的至高无上,法律只是皇帝自己颁布的制定法。换言之,没有对皇帝权力的司法制衡,遂给暴政留下充分余地。
对中国政治制度而言,这至少提出四个基本问题。第一,缺乏法治给政治带来的影响。西方有悠久的传统,把中国列作“东方专制主义”。这种想法是出于无知、傲慢和欧洲中心主义吗?或者,中国皇帝的确比西欧的君主掌握更大权力?
第二,中国制度中的合法性来自何方?中国历史充满无数起义、篡位、内战和改朝换代的尝试。然而,中国人始终返回平衡,让他们的君主掌控巨大权力,这样做的原因何在?
第三,尽管存在着周期性的皇权专制,中国统治者为何没有尽量行使理论上所享有的权力?虽然没有法律,他们的权力仍有实实在在的制衡;中国历史上有很长时期,皇帝主持稳定和守序的政体,没有肆意侵犯百姓的日常权益。还有很多时期,皇帝确实很弱,无法在刁蛮社会中强制执行规则。在传统中国,究竟什么在设置国家权力的真正极限?
最后,就仁政的性质而言,中国历史为我们提供何种教训?中国人发明了现代国家,但阻止不了国家的重新家族化。中国王朝历史的后续世纪就是一段持久的斗争史,防止这些制度的衰退,抵制权贵为自己和家庭谋求特权的权力家族化。什么力量促进政治衰败,以及它的逆转?
我将尝试在本章解答头两个问题,以下一章解答后两个。但首先得概述一下从唐朝到明朝的中国历史。
唐宋过渡之后的中国现代化
我最后一次讨论中国是在第 9 章。从 3 世纪到 6 世纪,中国经历了三百年的政治衰败。我们追踪它的发展,直到隋唐的重新统一。我提到,秦汉时期就已到位的现代国家制度,遭受严重的崩溃,政府重又家族化。汉朝之后的继承国,多半由贵族家庭掌控,他们将亲戚安插在主要职位,竞相攫取更多权力。重新统一中国的隋唐两朝的创始人,杨坚和李渊,都出自这个阶层。前者来自北周重要的贵族家庭,后者来自中国西北部的李氏望族,曾被封为唐国公。①像大部分继承国,隋朝和唐朝早期都操纵在贵族手中,他们官居要职,统帅军队,掌控地方政权。这个精英由北方军事贵族组成,其成员与鲜卑等野蛮血统进行广泛的通婚。605 年重新建立的科举制度,只是敷衍了事,在招纳非精英进入仕途上乏善可陈。②
唐朝持续近三百年,但在后期非常不稳定(请看表 2 的朝代排列)。从 7 世纪中期“邪恶”皇后武则天崛起开始,贵族精英杀死很多自己的同伴。到 8 世纪中期,帝国东北边境上的粟特—突厥(Soghdian-Turkish)将军安禄山发动叛乱,唐朝皇帝和太子不得不在深更半夜朝不同方向逃出首都长安。叛乱在八年后终告平息,但帝国中心区域的内战导致了人口的大量损失和经济衰退。帝国再也没有获得全盘恢复,权力流失到愈益自治的边境节度使。中国政治制度始终保持文官政府对军队的控制,但从此时开始像罗马帝国,强悍的将军将辖下的藩镇当作权力基础,追求自己的政治前程。唐朝最终在 10 世纪第一个十年中崩溃于叛乱和内战,北方出现军人掌权的五个短命朝代,南方则看到十个王国你方唱罢我登场。
表 2.后期中国朝代
| 年份 | 朝代 | 创始人/庙号 |
| 618 | 唐 | 李渊/高祖 |
| 907 | 后梁 | 李温 |
| 923 | 后唐 | 李克用 |
| 926 | 后晋 | 石敬瑭 |
| 947 | 后汉 | 刘知远 |
| 951 | 后周 | 郭威 |
| 960 | 北宋 | 赵匡胤/太祖 |
| 1127 | 南宋 | 赵构/高宗 |
| 1272 | 元 | 忽必烈 |
| 1368 | 明 | 朱元璋/太祖 |
| 1644 | 清 |
尽管有将近五十年的中断,中央国家的合法性在唐朝末年仍然获得广泛的认同,以致将领之一的赵匡胤在 960 年重新统一中国,以太祖皇帝的名号开创宋朝。在很多方面,宋朝在文化思想上是最多产丰饶的朝代。佛教和道教在隋唐两朝广受中国百姓和精英的欢迎,而儒家在北宋期间得到巨大的复兴,夺回不少信徒。宋明理学是一次强大的思想运动,波及邻国的朝鲜和日本,大大影响了整个东亚的思想文化生活。③
同时,中国开始承受一系列来自北方部落的入侵,他们得以占领大片领土,最终竟是整个国家。④边患始于契丹,它是蒙古边界的一个突厥—蒙古民族,在中国北方建立了庞大的辽国,夺得汉族聚居的燕云十六州。党项人在辽国西边创建了西夏,包括前几朝已受中国控制的边界地区。下一个出现的是来自东北的女真部落(满族的老祖宗),它击溃辽国,并把契丹赶到中亚。(他们向西逃得很远,竟然碰上俄罗斯人。自此,后者把所有中国人都叫作契丹斯基 Kitaiskiy。)1127 年,女真人洗劫宋朝首都开封,囚禁刚退位的皇帝和其儿子,迫使宋朝播迁南方,开创南宋朝代。女真人的金国在最旺盛时控制大约中国的三分之一,直到 1234 年败于另一入侵的游牧民族蒙古人。⑤占领中国北部之后,忽必烈可汗率领的蒙古军向西南发起进攻,一举占领整个中国。1279 年,蒙古军追逐南宋朝廷到广东沿海小岛的崖山。在蒙古军的团团包围下,数千朝臣自悬崖跳入海中自尽⑥,忽必烈可汗成为新创元朝的第一任皇帝。元朝统治者最终在 1368 年的民族起义中遭到驱逐,为本土的明朝所取代。
春秋战国时期的持久战争激发了愈演愈烈的建国举措,宋朝时的外敌入侵,却没对中国政治秩序发挥类似的作用。尽管有北宋兴起的理学派的辉煌成就,这仍是一段相当令人沮丧的时期,中国朝廷内部的派系斗争,阻止了政权对迫在眉睫的边患作出充分准备。军事压力来自社会发展程度远远低于中国的游牧民族,反而成为骄傲自满的理由。在当时的人类历史节点上,国家层次的社会与组成灵活骑兵的部落民族对峙,并不一定因先进的政治发展而取得决定性的军事优势。如阿拉伯哲学家伊本·赫勒敦所指出的,中国、中东和欧洲,因为邻接中亚辽阔的大草原,而遭遇周而复始的衰落—野蛮人征服—文明复苏。契丹、党项、女真和蒙古一旦征服中国领土后,最终都采用中国制度,走后也没留下重要的政治遗产。只有欧洲先进“野蛮人”前来征服,方才刺激中国政治制度酝酿更为根本的改革。
从隋朝开国的 581 年到 12 世纪的宋朝晚期,中国最普遍的政治发展之一是家族政府的逆转,中央集权得以复原到西汉的古典官僚制。到结束时,中国政府已不再受贵族家庭小圈子的控制,治理国家的是从社会广泛阶层招纳来的士绅精英。官僚作为儒家价值的监护人,其道德节操获得修复,并为 14 世纪明朝的可观政府打下基础。中国人口在这段时期急剧增加,到 1000 年已有五千九百万,到 1300 年更高达一亿。⑦中国开发南方的大片边境地区后,其领土也扩充到几近今日的版图。在这巨大的疆域上,随着运河和道路的建造,商业和通信获得实质性的增长。尽管疆域辽阔,中国还是发明了中央集权的政治制度,在错综复杂的社会中设定规则,征收税赋。统治如此广阔领土的欧洲国家,还要再等五百年。
中国建立(或重建)较为现代的政治制度,不是在 17 世纪和 18 世纪与西方接触之后,而是在唐宋之间的过渡期,这一见解首先来自第一次世界大战之后的日本新闻记者兼学者的内藤虎次郎(Naito Torajiro),即内藤湖南。⑧内藤认为,贵族统治在公元 750 年之后的动乱时期遭到席卷。其时,唐朝经历一系列叛乱和战争,非贵族背景的军事强人乘机掌权。宋朝在 960 年当政,皇帝不再受贵族家庭的威胁,形成更为纯粹的中央专制主义。科举制度成为选拔官员更为公开的途径,平民对贵族地主的农奴般的义务终告结束,其地位得到改善。共同的生活模式在全国建成,较少依赖世袭特权,白话文和平易近人的通俗文学和历史话本,逐一取代唐朝高度正规的文体。内藤从中找到与早期现代欧洲的显著平行,其时的欧洲,在强大专制国家的庇护之下,终止封建特权,引进公民平等。⑨虽然内藤的假设引起很大争议(尤其是他将西方分期法套用在东亚历史上的努力),但他的主要结论中,已有很多获得了晚近学者的认可。⑩
我们现可以返回本章开头的中国政治秩序的四个问题,首先是专制问题,中国的专制是否比其他文明中的更为严峻?
“毒侔蛇虺”的女皇武则天
被后世中国史家称为“毒侔蛇虺”的武曌(624—705),其故事值得在此重提,其意义不只是它可以告诉我们中国政治的性质。女皇武则天是以自己名义统治中国、并建立自己朝代的唯一女子。她的起伏是一部有关阴谋、残忍、恐怖、性、神秘、女人掌权的编年史。她是极具天赋的政治家,单凭自己的意志和狡猾而获得权力。儒家意识形态以歧视女子著称,在这样背景之下,她的成就显得格外刺目。⑪
我以前讨论法治时曾提到,它最初往往只适用于精英,而不是广大的民众,普通大众被认为不算完整意义上的人,不值得法律的保护。另一方面,在法治不存在的地方,精英成员通常比普通人面临更多危险,因为在上层赌注更大、权力斗争更激烈。这就是武则天治下的情形,她向中国的古老贵族家庭撒出恐怖的天罗地网。
有些历史学家,尤其是马克思主义的,在武则天的兴起中看到重大的社会启示。有的认为,她代表了上升的资产阶级;有的说,她是人民大众的斗士;还有的认为,她发挥了重要作用,把隋和唐初的家族精英赶走,代之以非贵族官员。尚不清楚,这些理论中哪一条最终证明是正确的。她自己拥有无懈可击的贵族血统,与隋朝皇族杨家有渊源。她并没有提携能干的平民,事实上她取消科举考试数年,为的是在官僚机构安插自己的宠臣。她对唐宋过渡的贡献,表现在她清洗实际上和受怀疑的贵族对手,大大削弱他们的人数,使整个贵族阶层变得孱弱,从而为安禄山的叛乱铺平道路。安史之乱标志唐朝走向末路的开始,促动了中国社会的巨大转型。
像中国宫廷的很多其他女人,武曌发迹于当上唐朝第二个皇帝太宗的低级嫔妃。她父亲是唐朝第一个皇帝高祖的拥护者,后来升任高职。如上所述,她母亲是隋朝皇室的后裔。据谣传,她与太宗的儿子高宗甚至在太宗去世之前就已有染。太宗死后,她削发为尼,搬到佛教寺庵。但新皇帝高宗的王皇后,想转移丈夫对淑妃的宠爱,故意将她带进宫,以观鹬蚌之争。
这证明是个致命的大错。高宗皇帝为武曌神魂颠倒,在他漫长的当政时期,证明自己是软弱的,很易受武曌的迷惑。武曌与皇帝生得一女,在无儿女的王皇后来访之后,设法让女儿窒息而死。王皇后被控杀死武曌的女儿,与淑妃一起被废成庶人,家人都被放逐到遥远的南方。随之,武曌获得晋升,到 655 年当上皇后,遂下令将王皇后和淑妃截去手足,投入酒瓮。曾支持王皇后、反对武曌为皇后的宫廷官员,包括曾忠实服务于前代皇帝的,或被放逐,或被处死。
很多中国女子躲在当上君王的儿子或丈夫的幕后,却行使实质上的大权,但武皇后决心变成真正的共同皇帝(编按:与高宗一同上朝,临朝听政,合称“二圣”),在公共场合中愈益显示自己的自主权。皇帝为了摆脱她的操纵,曾指责她玩弄巫蛊和妖术。但她当面力争,反而迫使皇帝杀死控告者,并从宫廷中清洗他们的拥护者。她恢复古代仪式,为自己和丈夫加封,震撼宫廷;为了逃避所谋杀的很多对手的鬼魂,她从长安迁都到洛阳。武皇后安排毒死自己身为太子的长子,诬蔑二子阴谋篡夺父位,将他放逐,迫他自杀。她丈夫最终于 683 年去世,她又把继承者(她的三子)中宗从皇位上拖下,处以幽禁。
不出意外,武皇后的兴起导致了 684 年的公开叛乱,叛乱来自身受其害的唐朝贵族家庭。武皇后迅速予以镇压,然后设置间谍和告密者的网络,厚赏检举者,从而对整个贵族阶层实施恐怖统治。她任用酷吏广泛从事现在所谓的“法外扑杀”(extrajudicial killings)。等恐怖发作完毕,她又把矛头指向酷吏头目,把他们也给杀了,这一切为她建立新朝铺平道路。690 年,她改国号为周,不再以她男性亲戚的名义,而以自己的名义单独执政。
武则天提倡爱民政策,减轻赋役,削减靡费的公共开支,扶助老弱病贫。她也推动为女子著书立传,延长对母丧的哀悼,封自己母亲为荣国夫人。她确实发动了一场社会革命,杀死大量在朝做官的唐代贵族和儒家学者。但她提拔的,不是有才能的平民干部,而是自己的宠臣和阿谀奉承之人,为此而特别放松相关考试和教育的标准。她统治的末期充斥着神秘主义、众多男宠(往往与她的宗教激情有关)、公开的贪污受贿,对于这些她并未试图加以遏制。几近八十岁的她,最终在政变阴谋中被迫让位;儿子中宗登基,改回唐朝国号。
武则天的行为在中国统治者中不算典型,后世的儒家卫道士申斥她是尤其恶劣的统治者。但作为暴君,对政权内的精英进行大肆的恐怖统治,她在中国不是第一个,也不是最后一个。多数的欧洲君主,其行事处世较守规则,但对治下的农民和其他平民,往往更加残酷。
武则天的兴起反而给中国女子掌权带来挫折,因为后来的文人学士将她当作女人干政只会坏事的例证。明朝皇帝在宫门上悬挂一块铁牌,告诫自己和继承者,时刻小心后宫女子的阴谋。后者不得不回到幕后,重新操起遥控儿子或丈夫的故技。⑫
天命
武则天试图攫取皇位,创建自己的新朝代,这引出中国君主一开始如何取得合法性的问题。托马斯·霍布斯在《利维坦》中认为,主权国家的合法性来自不成文的社会契约;在这份契约中,每个人放弃随心所欲的自由,以保障自己的生命权,否则就会面对“人人相互为敌的战争”。如果我们以“群体”替代“人”,很明显,很多前现代社会的运作就凭借这种社会契约,包括中国。人类愿意放弃大量自由,将相应程度的酌情权力授予皇帝,让他施政,以保障社会和平。他们宁愿这样,而不愿看到历史上一再出现的交战状态。其时,寡头强人一边彼此厮杀,一边尽情剥削自己的臣民。这就是天命的涵义,中国社会将合法性赋予具体的个人和其后裔,让他们享有统治百姓的独裁权力。
中国制度使人困惑的,首先不是天命存在与否,所有君主社会中都有类似的东西。它其实是程序问题:觊觎皇位者如何知悉他(在武则天的案例中就是她)已获得天命?一旦得到,其他觊觎皇位者如有机会为何又不来抢走(要知道皇帝享有巨大的权力和财富)?
前现代社会的统治者,其合法性可来自多方面。在狩猎采集和部落的社会,它通常是某种形式的选举的结果,参与的如果不是全体成员,就是主要氏族。或者,部落的长者开会来投票决定谁当领袖。在封建欧洲,某种形式的选举程序一直存活到早期现代。名叫三级会议(Estates General)或议会(Cortes)的机构,聚集起来开会,以批准新朝代的当政。这甚至发生于俄罗斯,1613 年将权力转给罗曼诺夫王朝,为取得合法性而召开了缙绅会议(zemskiy sobor,编按:俄语зе́мскийсобо́р)。
王朝合法性的其他主要来源是宗教。在基督教欧洲、中东和印度,有强大的宗教机构,既可将合法性赋予统治者,也可将之收回(如格里高利七世与神圣罗马皇帝的较量)。通常,这些宗教机构在政治当局的掌控之下,别无选择,只好确认。但在权力斗争时期,这些宗教权威又可通过授予合法性的能力,而发挥举足轻重的作用。
中国不同于其他文明,因为天命涉及的既不是选举,也不是宗教赋予的合法性。中国没有类似三级会议的机构,可供社会精英开会,以批准新王朝的创始人;也没有宗教等级制度可提供合法性。中国制度中没有超凡的上帝,天命中的“天”,不是犹太教、基督教和伊斯兰教中的神。此外,后三种宗教各有自己明确的书面规则。更确切地说,天命更像“自然”或“大道”,可被打乱,但必须返回平衡。此外,基督教皇或穆斯林哈里发,将合法性赋予国王或苏丹,但中国不同,它没有宗教机构可代表“天”来授“命”。⑬
改朝换代永远涉及合法性,因为新朝代上台往往通过简单的篡政或暴力。天命概念第一次出现于公元前 12 世纪的商周更替,周武王很明显从合法持有人那里夺得王位。在随后四千多年的历史中,中国经历了多次的改朝换代。不但有主要朝代,像秦、汉、唐、宋、明,还有无数小朝代,像汉朝崩溃后的三国,唐朝之后的五代。此外,有时中国分裂成众多区域,各有自己的朝代。
成为王朝创始人不需要社会先决条件。有的是前朝的贵族和高级官员,如隋唐的创始人。也有的是平民,如汉朝的刘邦和明朝的朱元璋。事实上,明朝开国皇帝一开始只是农家孤儿,幸免于饥荒和瘟疫,在佛寺里充任小沙弥,后来成为红巾军的将领。红巾军是一起宗教运动,聚集农民、强盗和投机者向地方当局的不公正提出挑战。自那以后,他在愈益澎湃的反元运动中统领越来越多的军队。元朝末年的中国沦落到一系列地方军阀手中,朱元璋就是其中之一。像很多其他的王朝创始人,在某种意义上,他证明自己是最能干最严厉的军阀,最终攀上顶峰。
在中国,是否胜者为王,败者为寇?天命是否只是军阀权力斗争的事后核准?这在很大程度上是正确的。一点也不奇怪,这个命题已有大批中国文献,如公元 1 世纪班彪的文章,解释为何有些统治者应得天命,而其他的却不值。但很难从这些文章中,提取一整套原则或程序,既能明确解说天命的授予,又不便在事后套在成功者的头上。⑭个别领袖的统治能否享有“朝代”的称号,往往要等很久才能得到历史学家的确认,从而使当时颇为可疑的政权赢得合法性。历史学家牟复礼(Frederic Mote)指出,默默无闻的北周创始人郭威和十年后创建强大宋朝的赵匡胤,他们都事涉篡位,上台都与背叛和欺骗有关,很难分辨。郭威的北周早早夭折,只因为儿子郭荣在三十八岁意外去世。如果郭荣活得长久,赵匡胤可能只是历史上一名试图搞叛国政变的能干将领。⑮
但皇帝和强悍军阀之间的道德距离还是非常遥远的。前者是合法统治者,他的权力得到大家的自愿服从,后者只是暴力的篡位者。哪些领袖有资格获得天命,哪些没有,中国精英自有一套理念,虽然不能付诸明确的程序规则。儒家的正名思想意味着,皇帝必须遵循理想前任的榜样,还必须拥有马基雅维利所谓的成功君主的美德。显而易见,未来皇帝必须是天生领袖,能激励他人追随自己的权威,敢于冒险以实现自己的目标。最常见的领导能力是指挥军事(武功),所以有很多王朝创始人都是以军事将领起家。但与其他文明相比,中国又比较不重视军事威力。儒家心目中的理想人选,是饱学的士大夫,而非粗野的军阀。觊觎皇位者,如果展示不出对儒家价值的恭敬和自身的教养素质(文治),便招揽不到宫廷内外各式派别的支持。牟复礼把明朝创始人朱元璋和他的竞争对手张士诚对照起来:
张士诚当过走私犯和强盗,在潜在的精英顾问和政治伙伴眼中,成了他的先天不足。很难在他的痞子经历中找到将会有大造化的证据……其早期谋士在他身上开了一个文人玩笑,朱元璋对此津津乐道。那些早期谋士给他和他兄弟换上雅致的大名,选了“士诚”二字,但没告诉他,《孟子》中有一名句,也包含依次出现的这两个字。但只要移动一下句读,该名句便变成:“士诚,小人也”。这一巧妙的蔑视让朱元璋哈哈大笑,直到有一天他怀疑,身边的文人顾问也有可能在用同样的妙计诋毁自己。⑯
中国的社会精英没有投票批准新朝代,但在潜在统治者的权力斗争中,仍发挥重要的幕后影响。天命并不总是授给最残忍最暴虐的军阀,虽然这样的人不时在中国上台执政。 很多像武后那样的觊觎皇位者,安排参与使自己获得君王权威的仪式——选择自己的庙号和朝代开始的年号——但很快垮台。中国制度能在建制化上做得特别讲究。一旦呈现某人拥有天命的社会共识,其合法性通常不会受到挑战,除非出现异常。在这一方面,中国的政治制度远比周遭的部落社会先进。
第 21 章 “坐寇”
所有国家都是掠夺性的吗?能否给明朝的中国贴上如此标签;中国历史后期的独断专行;没有对行政权力的制衡,能否维持清廉政府
经济学家曼瑟尔·奥尔森在一篇颇有影响的文章中,提出政治发展的一个简单模式。①世界最初落在“流寇”(roving bandit)的手中,像 20 世纪早期中国的军阀混战,或 21 世纪初在阿富汗和索马里的军阀割据。这些强盗纯粹是掠夺性的,经常在短时间向居民榨取尽可能多的资源,以便移往他处,寻找其他受害者。到一定时刻,其中一员变得鹤立鸡群,掌控整个社会:“这些暴力企业家当然不会自称为强盗,恰恰相反,他们会给自己和后裔冠上高贵的名号,有时甚至宣称享有神授君权。”换言之,自称合法统治的国王只不过是“坐寇”(stationary bandit),其动机与他所取代的流寇,没有什么差异。坐寇知道,如果不做短期的掠夺,反而向社会提供稳定、秩序和其他公共服务,让它在长远时期变得更加富饶,更能承担税赋,自己也就得到更多的收获。对受统治者而言,与流寇相比,这是一大进步。“流寇定居下来,向百姓提供政府服务,这出自他的理性自私。这理性自私将使他从社会中榨取最大化的资源,以供自己的享用。他将使用垄断的强制权力,攫取最大化的税赋和其他勒索。”
奥尔森继续指出,坐寇的最大化税率可与微观经济中的垄断价格媲美。如果实际税率超过这个限制,将打消生产动机,从而导致总税收的下跌。奥尔森认为,专制统治者不可避免总是制订最大化税率,而民主政权总是制订比专制政权更低的税率,因为它们必须求助于承担主要税责的“中间选民”(median voter)。
统治者就是坐寇,从社会中榨取最大值的税赋,除非在政治上受到阻止。奥尔森解说政府如何运作的这一概念,虽然愤世嫉俗,却讨人喜欢。这符合经济学家的努力,他们试图将理性的功利最大化行为模型推进政治领域,把政治看作经济的衍生物。这非常吻合美国政治文化的反中央集权的传统,后者对政府和征税始终保持怀疑态度。这还为政治经济学和政治发展理论,提供了预言性的漂亮模式,近年来得到了其他社会学家的极大扩展。②
但奥尔森理论是不正确的。传统农业社会的统治者,经常无法使用奥尔森的最大化税率向臣民征税。要回到一个不完全货币化的社会,凭借残缺不齐的历史税收数据,估算出当年的最大化税率,当然非常困难。但我们知道,前现代统治者经常增税,以满足像战争等的特定需求,待到紧急状态结束时再予以减税。仅在特定时期,统治者才会把社会逼上适得其反的绝境,这通常发生于朝代末期,以救燃眉之急。正常年代,他们向社会的征税一定远远低于最大值。
奥尔森模式的欠缺,最佳例证就是明朝中国。广泛的共识认为,当时的税率远远低于理论上的最大值,甚至低于最基本服务所必需的水平,譬如保障社会生存的国防。在明朝中国发生的,同样也会在其他农业社会发生,如奥斯曼帝国和欧洲的君主政体。这还可成为其他理论的组件,以解释传统政权为何很少采用最大化税率。③
皇帝并没行使理论上的权力,不单表现在征税上。武则天式的专制只是偶见,并不是持续现象。很多中国统治者对治下的百姓,表露出可被称为仁慈或忍让的态度,或儒家所谓的“仁”。中国有悠久的抗税历史,儒家的传统更认为,重赋代表了国家的道德缺陷。《诗经》就有如下的诗歌:
硕鼠硕鼠,无食我黍!三岁贯女,莫我肯顾。
逝将去女,适彼乐土。乐土乐土,爰得我所。④
明朝皇帝在权力上受到的约束并不来自法律。如我们在武则天的例子中所看到的,中国统治者不像欧洲统治者,如要增税,无须征求高等法院或议会的同意。他们不但可以颁发行政命令,任意调整税率,甚至可以随意没收他人财产。早期现代的法国和西班牙“绝对”君主,遇上强大精英时必须小心翼翼(参看第 23 章和第 24 章)。相比之下,明朝开国皇帝太祖,一下子就没收了全国最大几个地主的地产。据说,他清算了“无数”富裕家庭,尤其是在长江三角洲,因为他相信那里有特别顽固的反抗。⑤
对中国权力的真正约束大体有三种。第一,缺乏诱因来设置庞大的行政机关以执行命令,尤其是征收较高的税赋。明初,中国已是大国,其人口在 1368 年超过六千万,到 17 世纪末更增至一亿三千八百万。⑥在这样辽阔的领土上征税并不容易。在 14 世纪,货币流通很少,每个居民要缴的基本农业税都是实物⑦,通常是谷物,也可能是丝、棉花、木材和其他货物。当时没有综合的货币制度,以记录这些税赋,或将之转换成共同的计量单位。很多税赋归当地消耗(纳入预算),其余的运到逐级而升的粮仓,最终抵达首都(先在南京,后在北京)。纳税人承担的运输费用,往往超过所运货物的价值。地方和中央的收入和预算不做分门别类。有学者将之比作老式的电话接线板,电线来自各方,再插入各方,复杂得像一团乱麻。⑧户部人手不足,根本无法控制或理解这个制度。作为土地税基础的土地清查,实施于朝代早期,但并不齐全,之后又没有更新。人口增长、所有权变更、地理变化(洪水淹没或开辟荒地),很快使人口登记册过时。像其他民族,中国人也非常擅长于隐藏资产,并策划掩饰收入的计谋。⑨
皇帝征税和没收的无限权力常常是闲置的。它的使用多在朝代初期,皇帝正在巩固权力,与早先的对手一一算账。但随着时间的推移,宫廷经常需要那些精英的合作,便在早先没收财产的地区实施显著较低的税率。
第二,缺乏行政能力所限制的只是供应方面,而不同的皇帝也有自己不同的税收需求。奥尔森假定,任何统治者都想获得税收最大化。这反映了现代经济学的普通假设:最大化是人类行为的共同特征。但这是时代倒错,将现代价值向历史投射,当时社会并不一定同享这种价值。明朝开国的太祖皇帝是一名非常节俭的独裁者,他削减中央政府,避免涉外战争,粮仓实际上常有盈余。他的继承者明成祖朱棣(1360—1424)则截然不同,启动了雄心勃勃的营造运河和宫殿的大工程。明成祖也资助宦官将领郑和(1371—1435)下西洋,其巨型舰队抵达非洲,甚至可能更远。其政府开支是太祖时的两至三倍,额外税赋和徭役都有相应提高,引发了抗税起义和普遍不满。结果,第三任皇帝和后续继承者只好降低税率,向太祖时的水平靠拢,还向受触犯的士绅阶层作出其他政治让步。⑩明朝的大部分时期,土地税定在总产量的 5%,远远低于其他农业社会。⑪
中国君主一点也不逊于其他前现代社会的统治者,却往往展示出经济学家赫伯特·西蒙(Herbert Simon,中文名司马贺)所谓的“适可而止”(satisficing)行为,而不是最大化行为。⑫也就是说,如果没有如战争所引起的急需,他们经常满足于让睡着的狗继续躺着,仅仅征收应付正常需要的税赋。⑬下定决心的皇帝可能追求最大化税收,如明成祖,但所有专制政治领袖都会自动追求最大化的想法,显然不是真实的。
对皇帝权力的第三种限制不在征税和财政,而是权威的转授(delegation)。所有大型机构,无论是政府还是私人公司,都必须转授权威。这样做时,位居行政等级顶端的“领袖”,便会对机构失去相当程度的控制。转授的权威可以给功能专家,如预算官员或军队后勤,也可以给省、州、市和地方当局。这种权威转授是不可或缺的,因为统治者从来没有足够的时间或知识作出国内所有的重要决定。
权威转授的背后是权力转授。代理人以专门知识向委托人行使反制的权威。它可能是管理特殊部门的技术知识,也可能是某地区特别情形的本地知识。因此,像赫伯特·西蒙那样的组织专家认为,大型官僚机构中的权威不是一味从高到低,有时竟往往是反方向的。⑭
像现代的总统和首相,中国皇帝也遇上这类难题,官僚机构要么反应迟钝,要么蓄意违抗。尚书们或者反对皇帝的提议,或者悄悄地阳奉阴违。当然,中国统治者享有现代主管所没有的手段:他可以廷杖各级官员的赤裸屁股,或随便判以监禁和处决。⑮但这种强制方案,并没解决委托人和代理人之间潜在的信息问题。官僚经常不执行领袖的意愿,因为他们比较了解帝国的实际情形——并可欺上瞒下。
像中国那样的大国,其治理必须转授权力,必须依赖地方政府。不过,地方政府会滥用职权,腐化堕落,甚至共谋以反中央。正规的行政机构不足以对付此类问题。命令自上而下层层传达,但信息不一定回馈上去。如果他根本不知道滥权的发生,最独裁的皇帝也不会去惩治恣意妄为的官员。
君主权力的局限,曾在“封建制”和“郡县制”孰是孰非的标题下,在前现代中国受到讨论。这里的封建与欧洲封建主义的复杂内涵毫不相干,只表示权力的分散,而郡县制的地方官员都是中央指派的。根据明朝学者顾炎武(1613—1682):
封建之失,其专在下;郡县之失,其专在上。古之圣人,以公心待天下之人,胙之土而分之国。今之君人者,尽四海之内为我郡县犹不足也。人人而疑之,事事而制之,科条文簿日多于一日。而又设之监司,设之督抚,以为如此,守令不得以残害其民矣。不知有司之官,凛凛焉救过之不给,以得代为幸,而无肯为其民兴一日之利者。⑯
为了应付反应迟钝的行政机构,中国统治者的典型对策是设置间谍和告密者的平行网络,完全脱离正式政府,只是重叠在其上。这显示宦官所扮演的重要作用。不像普通官僚,宦官可以直访皇帝居所,通常获得比政府官员更大的信任。皇宫因此派他们外出,或刺探情报,或惩罚正式官僚。到明末,皇宫估计有十万宦官。⑰从 1420 年开始,他们组织成奥威尔式(Orwellian)的秘密警察,全名叫东缉事厂,受东厂掌印宦官的管辖,在朝代晚期演变成“极权恐怖主义的机关”。⑱但皇帝又发现,他也控制不了宦官。尽管有内正司(编按:明代负责惩处违纪太监的专门机构),他们还是自订政策,上演政变,共谋反对皇帝。⑲中国政治制度没有任何政治负责制的机制——没有地方选举或独立媒体,以保证官员的诚信。因此,皇帝不得不将一套自上而下的中央控制系统,叠放在另一套之上。虽然如此,他仍然无法取得对国家的严密控制。
明朝不愿和不能征收它所需要的税赋,最终导致它的倒塌。明朝统治的头两个世纪,中国基本上没有外患威胁。到 16 世纪末,安全情形急剧恶化。日本海盗开始突袭富庶的东南海岸,幕府将军丰臣秀吉在 1592 年侵犯朝鲜。同年,内蒙古发生战争,南方的土著也纷纷起义。最为严峻的是北方的后金,它变得更加强悍,组织得更加严密,已在东北边境频作骚扰。
政府对危机的回应完全无力。面临攀升的开支,它耗尽银子储备,但仍然拒绝向士绅阶层增税,最终坐失良机。虽然军事威胁变得愈益明显,累计欠税在 17 世纪最初几十年仍持续上升。皇帝甚至几次颁布税赋大赦,在征收欠税上显然认输。戍边军队早先组织成自给自足的军事屯垦区,现再也无法支撑,必须仰赖中央政府长途运来的给养。政府没能组织妥善的押运制度,因此做不到准时支付军饷。朝廷步履蹒跚,勉强维持到 1644 年。其时,北京政府因李自成的汉族起义军的打击而愈益衰弱,最终毁于获得明军降将帮助的满洲军队。
好政府,坏政府
20 世纪之前,明朝是统治中国的最后一个本土政权,其传统政治制度已发展到登峰造极的地步。它的机构现在看来是非常现代和有效的,但其他方面却落后和失灵得难以置信。
首先是帝国的官僚选拔制度。科举制度的根源可追溯到汉朝,但在隋、唐、宋初,出仕人选仍局限于精英家庭的小圈子。到了明朝,科举制度才成为进入政府的主要途径,赢得了威望和自主,使之成为所有后世科举制度的榜样。
科举制度与更广泛的教育机构相连。全国各地都有儒家学校,接受望子成龙的父母送来的孩子。最好的学生由老师推荐去南京和北京的国子监深造,将来参加科举考试。(推荐不争气学生的老师要受罚。此法现代大学可以借鉴,用以抵制贬值的分数。)精英家庭仍有可能以“例监”的名义,将自己的孩子送进去。但这些靠捐纳取得资格的监生(类似于当代哈佛和耶鲁的遗产特选生,即富裕校友的孩子),很少抵达官僚机构的最高层,那里仍然严格要求选贤与能。⑳最高荣誉属于连中三元者,即在三级考试中都获第一名:省的乡试、京城的会试、宫廷的殿试。在明朝历史中,完成此一壮举的仅商辂一人。他在官僚机构中级级高升,到 15 世纪晚期成为谨身殿大学士。㉑
中国的官僚机构树立了一个模版,几乎所有现代的官僚机构都是它的复制品。它有中央集权的委任和晋升制度,各等官职从顶端的一品到底部的九品(很像美国政府的文官序列表),每一品又分正从两级,所以,官职提升可从正六品到从五品。经科举而入仕的官员,会被派到全国各地担任低层官职,但不得在自己家乡。如果亲戚碰巧分配在同一衙署,年少的通常必须引退。三年之后,官员得到部门主管的评估,再直接上报吏部。不鼓励官僚的水平调动。经受住这个制度淘汰、并被提升到顶端的官员,往往是才华特别出众的。㉒
然而,这些才干优长、组织良好的官员在为一位独裁者服务。他无须遵守任何规则,大笔一挥便可否决仔细谋划出来的政策。他们面对皇帝变化无常的处罚和清洗,只有很少高官得以结束自己的任期,而没有受到羞辱。最坏的决策出自开国的太祖皇帝,他对自己的丞相产生怀疑,不但废除丞相制,而且规定“以后嗣君,勿得议置丞相,臣下此请者,置之重典”。这意味着,后代皇帝不能有相当于总理的助手,只好亲自与掌管实际工作的数十部门打交道。这个制度在精力充沛、巨细无遗的明太祖手中,尚能勉强运转;在能力较差的后代统治者手中,简直就是一场灾难。十天内,太祖必须应答 1,660 本奏章,处理 3,391 件不同事项。㉓可以想象,继任者对太祖所规定的工作量的愤慨。
很多后代皇帝不胜其任。传统上认为,明神宗(万历皇帝)是最不堪者之一。他自 1572 年到 1620 年的漫长统治,正好对应着明朝的式微。㉔在位的后半期,他干脆拒绝与尚书们见面和主持朝廷。数千份奏折留中不发,在宫廷里堆积如山,既不看也不予答复。事实上,他一连数年不出宫殿,其间重要的政府决策都无法制定。他也非常贪心,挪用国家财政来支付私人费用,例如建造壮观的定陵。17 世纪早期的军事危机中,国家储备仅剩二十七万两银子,他自己名下却累积两百多万两。不顾户部尚书的屡屡请求,他仍拒绝发放足够的帑银来支付军饷。㉕他的行动直接导致了最终摧毁明朝的满族力量的增长。
“坏皇帝”的问题
我们所讨论的政治发展三大组件中——国家建设、法治、负责制——中国在历史早期就获得了第一件。在某种意义上说,中国人发明了好政府。他们设计的行政机构是理性的,按照功能而组织起来,以非人格化标准进行招聘和晋升,这绝对是世界第一。也许因为中国社会如此重视家庭,国家建设者认定,他们的特别任务就是在政府中杜绝腐败根源的家族或裙带的影响。
在战国时期的战争洪炉中建立如此制度是一回事,要在后续两千年中维持下去是另外一回事。早已获得现代性的官僚机构,在国家崩溃或遭受贵族家庭的瓜分时,又变成衰败和家族制复辟的牺牲品。国家衰退在数世纪内逐渐发生,再要恢复到当初秦汉创建者的设计,也要花费数世纪。到了明朝,古典制度在很多方面获得完善。它更加任人唯才,所控制的社会比汉朝的更为庞大,更为复杂。
在其他方面,中国政治制度又是落后的。它从没创立法治和政治负责制的机制。国家之外的社会像以前一样,与欧洲或印度相比,组织得更为松散,很难采取政治行动。没有拥有土地的独立贵族,也没有独立城市。四下分散的士绅和农民,只可被动地抵制政府命令,不时爆发激烈的起义,又遭到残酷的镇压。他们从来没有像斯堪的纳维亚农民所做的那样,组织成集团向国家争取权利。随着佛教和道教的流传,独立的宗教团体在隋唐时期蓬勃兴起。在中国历史的不同时期,这些宗教团体发挥反国家的作用,从红巾军到太平天国。但宗教始终只是小宗派现象,在正统儒家当局的眼中是可疑对象,从没能代表强大的社会共识,也不能以法律监护人的资格来限制国家权力。
中国王朝的重大遗产是高品质的威权政府。世界上几乎所有成功的威权现代化者,包括韩国、新加坡,现代中国大陆、台湾地区,都是分享中国共同文化遗产的东亚国家,这不是偶然现象。很难在非洲、拉丁美洲或中东,找到像新加坡的李光耀或韩国的朴正熙那样素质的威权统治者。
但明朝和中国其他历史时期的经验,提出一个令人不安的问题:在没有法治或负责制的情况下,良好统治能否长久。如遇坚强能干的皇帝,该制度卓有成效,雷厉风行,简直令人难以置信。如遇变化无常或庸碌无能的君主,他们大权独揽,经常破坏行政制度的效率。武则天清洗官僚机构,安插自己不合格的追随者;明太祖废除丞相制,让继任者束缚于这一困境;明神宗完全不理政事,导致政府瘫痪。中国人视之为“坏皇帝”问题。
中国制度中确有一种负责制。皇帝接受教育,深感对人民的责任。他们中的优秀者,尽量回应人民的需求和抱怨。尽责的统治者还经常以人民名义惩戒手下官员,并依靠宦官网络来刺探谁在做好事,谁在做坏事。但制度中唯一正式的负责制是向上的,即对皇帝负责。地方官员必须担忧,宫廷如何看待他们的表现,但绝对不会在意普通老百姓的意见,因为后者无法依赖司法或选举的程序来反对自己。对普通中国人而言,遇上昏官的唯一求援是上诉,希望皇帝有可能获悉。即使是好皇帝,在如此辽阔的帝国中,要想得到他的注意简直是缘木求鱼。
……
然而,法治和政治负责制在中国是不存在的。滥权的绝大多数,并不来自暴政的中央政府,而是来自散布四方的各级地方官员。他们狼狈为奸,或偷窃农民的土地,或接受商人的贿赂,或漠视环保和安全的规则,或遵循历来地方官员所从事的。如有灾难发生,例如地震披露的豆腐渣学校工程和管理不善的公司的奶粉污染,中国人的唯一求援就是向中央政府上诉。而中央政府则不一定作答。有时,它会对犯法官员采取严厉措施,但在其他时候,它自己太忙,或心不在焉,或要应付更为紧要的事务。
法治和政治负责制本身很好,但有时会搅乱卓有成效政府的运作,如印度国家由于诉讼和公众抗议,而无法作出基建项目的决策;或美国国会由于说客和利益团体,而不愿面对像社会福利这样的紧迫问题。
但在其他时候,为维护卓有成效的政府,法治和负责制又属必不可少。在适当条件下,强大的威权制度可以建立非常有效的政府。政治制度要能承受外部条件的变化,以及内部领袖的变更。法治和负责制制衡国家权力,从而减少政府表现的参差不齐。它们约束最好的政府,但也防止坏政府的失控。相比之下,中国人从未能解决坏皇帝的问题。
光有制度还不够
传统中国为何发展不出本土的资本主义?这引起了广泛争论,包括马克斯·韦伯的《中国的宗教:儒教与道教》和李约瑟(Joseph Needham)的巨著《中国的科学与文明》。本卷目的不是为了参与争论,只是想解说,遏制资本主义在中国发展的大概不是由于良好制度的缺席。
现被认为与现代经济发展休戚相关的制度,明朝中国已拥有大部。它有强大和组织良好的国家,可提供稳定性和可预测性。卖官鬻爵和其他公开的腐败虽然存在,但不像 17 世纪的法国和西班牙(参看 23 章和 24 章)那么猖獗。㉖暴力处于控制之中,与很多当代发展中国家相比,中国实现了文官政府对军队的高度控制。其弱点当然是法治的缺乏,产权因此而受害于政府的朝令夕改。如我在第 17 章中所争论的,对经济增长而言,宪政意义上的法治并不是必须的。虽然土地不时被征用,尤其是在朝代初期,但国家得以维持几十年“足够好”的产权,在农村的征税也尤其偏低。今天的中华人民共和国,也有足够好的产权,以支持异乎寻常的经济增长。㉗
当然,明朝中国奉行经济上不理性的政策,严格控制商人和贸易。它对食盐生产的垄断将价格人为提高,像法国和奥斯曼帝国一样,导致大量走私和腐败。对发展来说,政策远远没有制度那么重要,朝令可以夕改,而制度的建立则艰难得多。
中国所缺乏的,恰恰是经济学家假设为人类共同特征的利益最大化精神。明朝中国的各行各业,都沉浸在巨大的满足之中。皇帝觉得没有必要收取力所能及的税赋,其他种类的革新和变更也都不值一试。下西洋总兵正使郑和远航印度洋时,发现了全新的贸易通道和文明社会,但没有激起好奇心,也没有后续的远航。下一个皇帝为了节约而削减海军预算,中国的大发现时代(Age of Discovery)刚刚开始,便告结束。同样,名叫苏颂的宋朝科学家发明了世界上第一座机械时钟,由水轮推动庞大多层的齿轮系统,因女真人攻陷首都开封而遭遗弃。时钟的部件散落各地,如何制作,乃至它的曾经存在,经过几代人就湮没无闻了。㉘ 阻碍明清中国取得经济增长的因素,今天已不复存在。早期西方评论家认为拖中国后腿的文化缺陷,现也不再是原因。20 世纪初,大家都嘲笑儒家理想中的士绅学者,留长指甲,除了当官,拒绝做任何其他工作,成为现代化的障碍。这一独有的士绅理想已在 20 世纪消失,但重视教育和私人进取的文化遗产仍然生龙活虎,非常有利于中国的经济增长。它体现在全世界无数中国母亲身上,省吃俭用,把孩子送到最好的学校,敦促他们在标准化考试中出人头地。导致明成祖的继任者取消远航的自满,已被异乎寻常的强烈意愿所取代,中国领导人渴望学习外国经验,如果合适便加以采用。首创门户开放的政治家邓小平说,“不管黑猫白猫,捉住老鼠就是好猫”。中国在前一世纪全球经济比赛中表现得如此糟糕,现在又如此杰出。较为信服的解释是它对科学、知识和革新的态度,而不是它的政治制度的根本缺陷。
第四部分 负责制政府
第 22 章 政治负责制的兴起
何谓政治负责制;欧洲建国的迟到反成自由的来源;辉格史观错在何处;比较各国才能理解政治发展;欧洲五种不同的结果
负责制政府意味着,统治者相信自己应对治下的民众负责,应将民众利益置于自身利益之上。
负责制可以多种方式获得,如道德教育,这是中国和受儒家影响国家所奉行的。君主接受教育,深感对社会的责任,并从老练通达的幕僚那里,接受经邦纬国的咨询。今天,统治者自称关心民众,但又不受法治或选举在程序上的限制,如此的政治制度,西方人士往往嗤之以鼻。但道德负责制在威权社会中仍有实际意义,约旦哈希姆王国与萨达姆·侯赛因治下的伊拉克复兴党(Ba’athist)政权形成明显的对照。它们都不是民主政体,但后者实施残酷和无孔不入的专政,主要为萨达姆亲朋好友的利益服务。相比之下,除了权力极其有限的议会,约旦国王无须对人民负责,但还在尽量满足约旦社会各团体的需求。
正式的负责制只是程序上的:政府愿意屈服于限制其随心所欲的机制。归根结蒂,这些程序(通常在宪法中得到详细说明)允许社会公民因政府渎职、无能或滥权而将之完全取代。今天,程序上负责制的主要形式是选举,其中最好的是成人普选的多党选举。但程序上的负责制并不局限于选举。在英国,对负责制政府的早期要求是以法律名义,公民相信国王也应服从法律。其中最重要的是普通法,基本上是由非民选法官所塑造,再加上非普选议会所制订的。所以,最早形式的政治负责制,其对象不是全体人民,而只是代表社会共识的传统法律,以及寡头的立法机关。我在此使用“负责制”,而不用“民主”,道理就在这里。
久而久之,民主渐渐发生。选举权逐一抵达更为广泛的阶层,包括无产男子、女子、少数种族、少数民族。此外愈来愈明显,法律不再依据宗教,而要求得到民主的批准,即使其执行仍留给专业法官。在英国、美国和西欧,程序上负责制的完全民主化,一直要等到 20 世纪。
姗姗来迟的欧洲建国
早期现代时期,欧洲国家建设者方才投入等同于中国和土耳其的工程——建造强大的中央国家,在全国领土上实施统一的行政管理,并宣称主权。这些努力开始得很晚,始于 15 世纪末,成于 17 世纪末。国家主权的理论来自学者的笔尖,如格劳秀斯(Hugo Grotius)和霍布斯。他们主张,真正享有主权的不是上帝,而是国王。
总的来说,欧洲君主在此项工程中遇上更大阻力,与中国或土耳其相比,欧洲社会中其他政治参与者组织得更为严密。国家建设继续进行,但经常遭遇有组织的反抗,迫使统治者寻找同盟以求折中。地主贵族早已根深蒂固,坚守在固若金汤的城堡,拥有独立的收入和军队。中国贵族从未获得如此的独立;如我们所知,奥斯曼帝国从不允许此种贵族阶层诞生。国家建设广泛开展时,西欧涌现了资本主义经济的元素。商人和早期制造商创造大量财富,不受国家的控制。自治城市愈益成熟,尤其在西欧,还依据自己的规则来组织自己的民兵。
欧洲法律的早期发展在限制国家权力上发挥重要作用。君主经常侵占百姓的产权,但漠视法律依据而随意没收私人财产的却很少。因此,他们并不享受无限的征税权力,为了资助战争还要向银行家借钱。就任意的逮捕或处决而言,欧洲贵族享有更多的人身安全。除了俄罗斯,欧洲君主也避免在自己社会中向精英发动赤裸裸的恐怖和威胁。
欧洲国家建设的迟到,恰恰是欧洲人后来享受的政治自由的来源。早熟形成的国家,如果缺乏法治和负责制,能对百姓实施更为有效的暴政。物质条件和技术的每一项进步,落在不受制衡的国家手中,便意味国家更有能力为自身目的而严格控制社会。
向平等进军
托克维尔(Alexis de Tocqueville)在《论美国的民主》中开门见山:过去八百年中,人人平等的思想在世界各地得到认可,这一事实是天赐的(providential)。①贵族的合法性——有人生来就高贵——不再是理所当然。没有奴隶的改变意识和寻求承认,主子和奴隶之间的关系就无法颠倒过来。这一思想革命有很多来源。所有的人,尽管在自然和社会的层次有明显差异,但在尊严和价值上却是平等的。这个概念是基督教的,但在中世纪教会的眼中,其实现并不在今生今世。宗教改革,加上印刷机的发明,赋予个人阅读圣经和追求信仰的权利,不再需要像教会那样的中介。始于中世纪晚期和文艺复兴时期,欧洲人已开始质疑既存权威,现在这种质疑得到进一步的加强。那时,人们开始重新学习古典文献。现代自然科学——从大量实证数据中提炼普遍规则,通过可控试验来测试因果理论——树立了新式权威,很快在各大学中获得建制化。它所孵化的科学和技术,可供统治者利用,但不受控制。
奴隶日益意识到自己的价值而变得理直气壮,这种转变表现在政治上,就是追求自己的政治权利。换言之,他们要求分享共同决策权。该权利曾存在于部落社会,只因国家兴起而湮灭。这项追求导致了社会团体的大动员,像资产阶级、农民和法国大革命中的城市“群众”,曾经都是治下的消极老百姓。
这项追求寓于普世的字眼之中,对现代负责制政府的兴起至关重要——如托马斯·杰斐逊在美国《独立宣言》中所宣告的,它是基于“人人生而平等”的前提。纵观人类历史的先前阶段,不同个人和团体为获得承认而斗争,但其寻求的承认是为他们自己、他们的亲戚团体和社会阶层;他们试图自己成为主人,而从不质疑主子和奴隶的关系。对普遍权利的新式理解显示,接踵而至的政治革命,不再以新的狭窄精英团体去替换旧的,而在为全体人口逐渐获得选举权而铺平道路。
思想变化的累积效果是极其巨大的。法国有中世纪机构三级会议,如有国家大事,可召集全国代表来开会作出决定。1614 年,玛丽·德·美第奇(Marie de Medicis)摄政王召开的三级会议,对腐败和税赋频发牢骚,怨声载道,但最终还是接受皇家的权威。到 1789 年,由于启蒙和人权思想的影响,它的再次召开遂激发法国大革命。②
如果没有权力和利益的潜在平衡,使参与者认为它是糟糕选择中最好的,单凭思想观念,还不足以建成稳定的自由民主政体。强大国家既执行法律,又受法律和立法机关的制衡,这种奇迹全靠社会上不同的政治参与者彼此之间维持大致的均势。他们当中,谁也不是龙头老大,便不得不达成妥协。我们所理解的现代立宪政体,就是这些不受欢迎、计划之外的妥协的结果。
自共产主义倒塌和亨廷顿的第三波民主化以来,我们目睹了这种动态。第三波始于西班牙、葡萄牙和土耳其在 20 世纪 70 年代的民主过渡;到 70 年代和 80 年代,再转移至拉丁美洲和东亚;随着 1989 年后东欧共产主义的倒塌而抵达顶峰。民主政体是最为合法的,甚至是唯一合法的,这种思想已传遍世界每一个角落。民主宪法在非洲、亚洲、拉丁美洲和前共产主义世界获得重订,或首次制订。但稳定的自由民主政体,仅占参与民主过渡国家的一部分,因为社会力量的对比,未能迫使不同参与者达成宪政上的妥协。这个或那个参与者——通常是继承了行政权威的——总会比其他参与者更为强大,并以他人为代价扩充自己的势力。
支持现代民主的启蒙思想在欧洲广泛传播,一直抵达俄罗斯。各国接受程度则有显著的差别,取决于不同政治参与者对自身利益所受影响的估量。要了解负责制政府的出现,必须了解欧洲各地既存的政治力量,有些提倡负责制,另一些并不反对专制主义的抬头。
仅了解一个国家等于不懂国家
我谈论欧洲时,好像它是与中国或中东作比的单独社会,但在事实上,它拥有政治发展的多种模式。现代宪政民主的故事经常基于胜利者的观点,即老是依据英国和其殖民衍生品美国的经验。在所谓的“辉格史观”(Whig history)中,自由、繁荣和代议政府的同步成长,被视为人类制度无可阻挡的进步,其始于希腊民主和罗马法律,铭记于大宪章,虽受到斯图亚特王朝的威胁,但在英国内战和光荣革命期间,获得了捍卫和昭雪。这些制度通过英国在北美的殖民地,再输给世界各国。③
辉格史观的问题,不是指它的基本结论是错的。实际上,强调征税在驱动负责制政府出现上的首要作用,大体是正确的。问题在于,像所有仅从单一国家历史出发所作的论证一样,它不能解释议会制度为何出现于英国,而缺席于情形相近的其他欧洲国家。这种史观经常导致评论家断定,已然发生的事必然发生,因为他们不清楚导致特别结果的复杂背景关联。
举例说明,在兰尼米德七年之后的 1222 年,皇家侍从阶层迫使匈牙利国王安德鲁二世(Andrew Ⅱ)签署让步的金玺诏书(Golden Bull),被誉为东欧的大宪章。该诏书保护精英免受国王的随心所欲,如果国王违诺,主教和议会要员享有抵制权利。但这诏书从没成为匈牙利自由的基础。这部早期宪法在限制匈牙利国王权力上颇为有效,实际统治权竟而落到了不愿自律的贵族阶层手中。该宪法并没开发新政治制度,以立法机关来制衡行政权力,反而阻碍了强大中央政府的出现,以致国家无法抵抗外来侵略。国王也无法保护国内农民免遭寡头的贪得无厌。到了 1526 年的莫哈奇战役,匈牙利完全丧失自由,成为奥斯曼帝国的战利品。
负责制政府兴起的任何解释,既要看成功案例,也要看不成功的。这样才能了解,为何代议制度出现于欧洲某地而专制主义却盛行于其他地方。从德国历史学家奥托·欣策(Otto Hintze)开始,已有人在作出努力。查尔斯·蒂利再接再厉,认为外部军事压力和征税能力是主要的变量。④最近的卓越努力来自托马斯·埃特曼(Thomas Ertman),他查阅的案例远远超过大多数比较历史研究,并对大部分观察到的差异作出了较为信服的解说。⑤
这种研究还无法成为政治发展的真正理论。说到底,能否创立这样理论都还是未知数。从社会科学的角度看,麻烦在于有太多变量,而没有足够案例。该理论尝试解释的政治结局,不仅是代议政府和专制主义的黑白之分。如下所述,至少有五种不同类型的国家在欧洲出现,其起源都需要得到解释。例如,法国和西班牙的专制主义,跟普鲁士和俄罗斯的就相当不同。事实上,普鲁士和俄罗斯彼此之间又有很大差异。有实证显示,发挥作用从而导致不同结局的变量,其数字是很大的,既有蒂利说的外部军事压力和征税能力,还有内部阶级关系的结构、国际谷物价格、宗教和思想、统治者和民众接受变量的方式。要想从这么多因果关系中,找出可预测性的普遍理论,其前景确实微茫。
我将在后续章节中,尝试描述欧洲政治发展的重要路径,以及与此相关的各种原因。也许可从一系列案例中概括出哪些因素最重要哪些最不重要,但远远不能成为真正的预测性理论。
欧洲的东周时期
在很多方面,1100 年的封建欧洲很像周朝的中国。有名义上的君主或统治朝代,但实际权力落到高度分散的封建领主手中。他们保持军队,维持秩序,主持正义,在经济上基本上自给自足。也像中国一样,有些王室凭借严密的组织能力、冷酷无情以及运气,而变得出类拔萃,并开始在愈益扩展的地域中巩固自己的领土。
15 世纪到 17 世纪,欧洲发生巨大的政治变动,导致强大国家的兴起,可与中国公元前 5 世纪到公元前 3 世纪的国家建设媲美。变更背景是人口的大幅增长,尤其是在 16 世纪,再加上人均财富的递升。这是一个全球现象,如我们以前讲到的,也影响奥斯曼帝国。它在欧洲造成的效果,比在中东也许更为良性。欧洲人口从 1500 年的六千九百万,增至 1600 年的八千九百万,增长率几近 30%。⑥大量金银来自西班牙在新大陆的殖民地,经济货币化在迅速流行。贸易增长开始超过国内生产总值的增长,从 1470 年到 19 世纪初,西欧商船的规模增长十七倍。⑦
这段时期的一开始,多数欧洲政体只是“领地国家”(domain states)。国王的全部收入来自自己的领地,只占他名义上统治疆土的一小部分。行政人员很少,来自国王家庭。实际权力分散在各级封建属臣手中。他们都是自治的政治实体,保持自己的军队,向自己的百姓征税,在地方上主持正义。如果自己是强大的男爵,就提供服务给国王。如果自己是较低等级的属臣,就提供服务给男爵。他们不是以税赋而是以自己的鲜血来履行义务,或亲自披挂上阵,或率领侍从。事实上,大多数贵族因此而免缴税赋。国王的领地可能散播于辽阔的疆土,分成数块,互不相连。他的王国只是各级属臣领土的拼凑图,甚至忠于敌对国王的属臣也会间杂其中。
到这段时期结束,大部分欧洲政治秩序已转化成国家体系。领地国家转化成缴税属国,君主的收入不仅来自国王自己的领土,而且来自他所能征税的整个疆域。管理这个制度需要更大的国家官僚机构,最开始是秘书处和财政部,以掌控收入的征集和支付。地方领主的自治受到严重限制,现在需要缴税,而不再提供服务。中央政府向农民直接征税,从而破坏了领主与农民的传统关系。欧洲教会的地产都被国家夺走,国家直接控制的领地显著增加。国家司法的领土也从互不相连的拼凑图,变换成相邻的一整片。例如,法国版图就是在那时形成现在熟悉的六边形。通过征服、联姻或外交,各国吸收弱小政治体而得以扩展。各国也开始渗透社会,以宫廷语言来统一和减少各地方言,调整社会习俗,在愈益增大的管辖区内,建立法律和商业的统一标准。
该变化的速度和程度颇不寻常,在很多方面可与东周时期的中国媲美,不同处只在最终幸存国家的众多,而不是大一统帝国。以征税为例,在哈布斯堡帝国内,1521—1556 年的征税为 430 万弗罗林(Florins),1556—1607 年便涨到 2,330 万。英国的平均年度税收,从 1485—1490 年的 5,200 英镑涨到 1589—1600 年的 382,000 英镑。卡斯提尔王国(Castile)在 1515 年征税 150 万枚达克特(ducat)金币,到 1598 年征税 1,300 万枚。⑧增加的税收用来支付更大更为专业的公共机构。1515 年,法国有七至八千官员为国王服务;到 1665 年,皇家行政人员升至八万。巴伐利亚政府在 1508 年有 162 名官员领取薪俸,到 1571 年增至 866 名。⑨
欧洲国家的早期发展植根于主持正义的能力,但到 16 世纪之后,几乎全是为了资助战争。这段时期的战争愈打愈大,几乎持续不断。其中大型的包括:法国和西班牙之间为争夺控制意大利的持久战;西班牙征服荷兰联合省的努力;英国、西班牙、葡萄牙、荷兰和法国在新大陆争夺殖民地;西班牙试图侵略英国;宗教改革之后日耳曼内的持续对峙(以三十年战争而告终);瑞典向中欧、东欧和俄罗斯的扩张;奥斯曼、哈布斯堡和俄国之间的战火连绵。
早期现代的国家除了基本治安和正义,没有提供多少服务。它们预算的大部用在军事开支。荷兰共和国预算的 90%,花在与西班牙国王的长期战争上。哈布斯堡帝国预算的 98%,用来资助与土耳其和 17 世纪新教政权的战争。17 世纪从头到尾,法国的预算上涨五到八倍。从 1590 年到 1670 年,英国预算增加了十六倍。⑩法国军队人数从 13 世纪的一万二千,增至 16 世纪的五万和 17 世纪 30 年代的十五万,再增至路易十四统治晚期的四十万。⑪
法律在欧洲发展中的作用
公元前第一个千年的中期,中国从少量贵族驾驶战车的战争,过渡到向全民征募的步兵战争。在 12 世纪和 13 世纪,类似的技术过渡也在欧洲发生,披甲戴盔的骑兵由配备弓矛的大批步兵所取代。跟中国的早期建国者不同,早期现代的欧洲君主没在自己领土上征募大量农民。查理五世(Charles V)投入战场的精锐军队,以卡斯提尔部队的步兵方阵(tercio)为核心,再配以来自国内外签有合同的雇佣兵。⑫欧洲的大规模征募仅出现于 18 世纪,但他们仍然不是国家权力的基础,直到法国大革命的国民征兵制(levee en masse)。相比之下,像秦国一样的东周列国,直接从骑兵的贵族战争过渡到大规模征募,中间没有雇佣兵阶段。⑬
早期现代的欧洲君主为何没像中国君主那样,直接征募自己领土上的大量农民?为何不以增税来付军饷,反而要依赖贷款和卖官鬻爵?
主要原因之一是欧洲的法治。我们在第 18 章中看到,它由宗教法律发展而来,在各领土上广泛流传。欧洲封建主义的整个等级结构,受到承继下来的法律的保护,将主权和权力有效地分配给各式从属政治体。农民受一系列封建法律和义务的束缚,主要是欠自己领主的。国王没有征募农民的法律权利,事实上,他甚至不能征募自己领土上的农民。因为后者的义务定得十分详细,可能没有军事服务。欧洲君主并不觉得自己可攫取精英的财产,因为后者可援引基于封建契约的古代权利。国家可以征税,但必须通过组织起来的各式会议(像法国的三级会议),以证明征税的正当性,方可取得许可。专制君主曾尝试削减这些会议的权力,但其操作仍局限于赋予君主合法性的法律总框架。国王并不觉得自己有权侵犯对手的私人安全,或任意拘留,或随便处死。(但要注意,这些规则很少用于非精英者,像农民和其他平民,他们还要再等到历史的后期。)
早期中国君主所实使的暴政,很少欧洲君主敢于尝试,不管是在封建时期还是早期现代。中国君主从事大规模的土地改革,任意处决当朝的行政官员,迁移整个区域的人口,疯狂清洗贵族对手。出现此类行为的唯一欧洲宫廷是俄罗斯。这种不受节制的暴力要在法国大革命之后,方才变得流行。当时,源于古老欧洲秩序的所有法律约束,被现代化一扫而空。
欧洲的国家发展必须应付限制国家权力的全套法律,懂得这一点很重要。欧洲君主试图扭曲、违反和回避有关法律,但其选择仍受成熟于中世纪的既存法律的限制。
国家建设的架构
为了投入战争,国家必须以愈益增大的规模动员资源。对资源的需求,导致更高水平的征税,想方设法将更多人口和社会资源纳入征税范围。财政资源的管理,促使国家官僚机构的扩大和机构的愈益合理化,以谋求最高效率。国家要有辽阔领土,以扩大税收基础;要有相邻领土,以达防御目的。政治异见会被敌人利用,因此有必要在整片领土上实施统一的行政管理。
欧洲的某些地区——日耳曼和东欧的一部分,还有像瑞士那样的地理隔离地区——没有面对早期的军事竞争,因此组织现代国家较晚。所有的其他强国——法国、西班牙、英国、荷兰、瑞典、俄罗斯、哈布斯堡帝国、波兰、匈牙利等——从 15 世纪以来,都面对军事开支和中央集权的需求。⑭
欧洲历史此时的政治发展,体现在集权国家和抵抗团体之间的互动。如果抵抗团体单薄且组织不良,或被国家收买去帮助榨取他人的资源,那里就出现专制政府。如果抵抗团体组织良好,中央政府无法颐指气使,那里就出现较弱的专制政府。如果抵抗团体与国家不相上下,那里就出现负责制政府,他们坚持“无代表即不纳税”的原则:愿意提供实质性的资源,但一定要参与如何使用的决策。
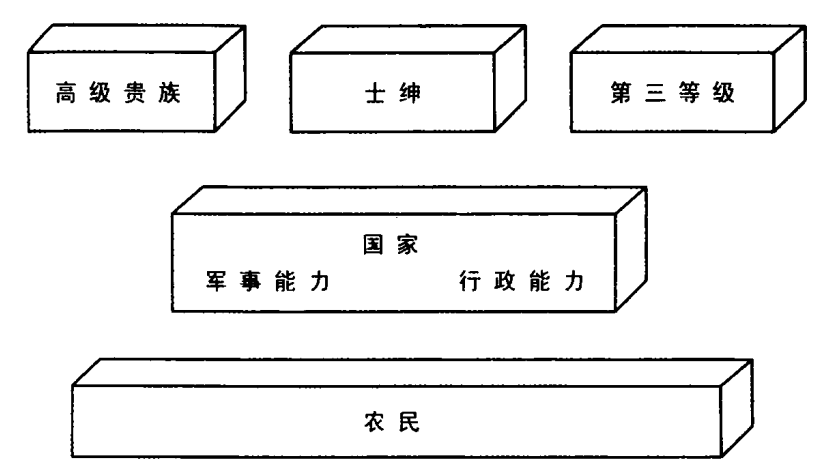
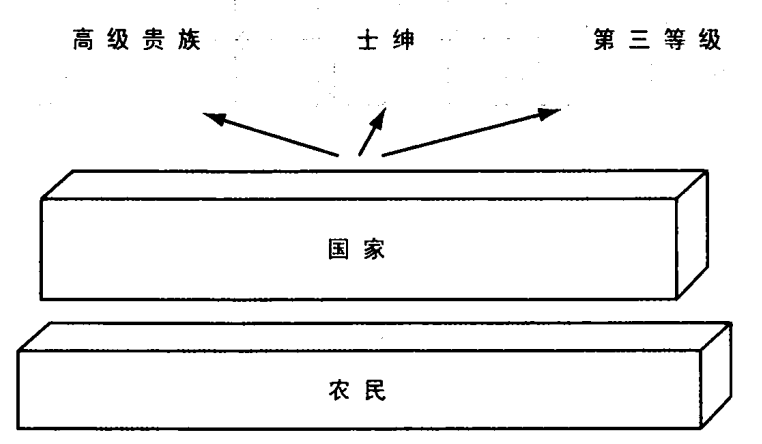
斗争的结果不是国家与整个社会的双边权利争夺战。粗略而言,斗争牵涉四支力量:中央君主政府,高级贵族,更为广泛的士绅阶层(小地主、骑士和其他自由人),包括市民在内的第三等级(资产阶级的雏形)。占社会人口大多数的农民尚不是重要参与者,因为他们还没动员起来,还没成为代表自己利益的社会集团。
对国家集权的抵抗程度,取决于国家之外的三个群体——高级贵族、士绅、第三等级——能否合作,以对抗皇家权力。它也取决于每个群体所显示的内部凝聚力。最终,它还取决于国家本身的凝聚力和使命感。
在后续章节中,我将显示四个欧洲国家建设的结果,以及这些结果为何迥然不同的原因。这个分类覆盖了最为纷纭的案例,从最为代议的到最为专制的。它们是:
1.软弱的专制:16 世纪和 17 世纪的法国和西班牙君主政体,代表了新型的专制国家,在某些方面,比荷兰和英国更为集权,更为独裁。另一方面,它们仍不能完全支配社会上的强大精英,更重的税赋落到了最无力抵抗的阶层。它们的中央政府仍是家族的,事实上,其家族制的程度日益增长。
2.成功的专制:俄罗斯君主政体收买贵族和士绅,使之变成完全依赖国家的服务阶层。能够这样做,部分原因在于三方都有共同利益,都想将农民绑在土地上,并向其征收最重的税赋。当时的政府仍是家族制的,但阻止不了俄罗斯君主对贵族阶层的恐吓和控制,其程度远远超过法国或西班牙国王所做的。
3.失败的寡头制:匈牙利和波兰的贵族一开始就向国王权力施以宪法限制,导致后者一直软弱,无法构建现代国家。软弱的君主政体无法保护农民利益,以对抗贵族阶层的残酷剥削;也不能提取足够资源来建造国家机器,以抵御外来侵略。这两个国家都没建成非家族的现代政府。
4.负责制政府:最后,英国和丹麦发展出了稳定的法治和负责制政府,同时又建成能发起全民动员和防御的中央国家。英国如何发展议会制度,这是耳熟能详的故事。斯堪的纳维亚通过不同的政治进程,却获得同样结局。到 19 世纪末,一个成为自由派国家,另一个奠定了社会民主主义国家的基础。法律和负责制的原则早已深植人心。
除了上述这些,还有其他重要的变量和结局。荷兰共和国和瑞士联邦,代表了另一类通向负责制政府和法治的共和途径。普鲁士君主政体虽然没有负责制,却发展了法治和强大的现代国家。我无法一一介绍这些和其他边缘案例。重要的是弄清大致的相关条件,哪些支持负责制政府,哪些支持不同形式的专制主义。
第 23 章 寻租者
法国的财政危机导致家族政府的兴起;总督和中央政府的成长;法国精英把自由当作特权,遂无法付诸集体行动;法国政府的致命弱点,既无法控制自己的精英,也无法向其征税
法兰西王国呈现极端矛盾的形象,既强大,又充满潜在的虚弱。参观过巴黎郊外凡尔赛宫的人都会明白,路易十四时代的欧洲人为何如此敬畏法国君主政体。相比之下,腓特烈大帝(Frederick the Great)在波茨坦的无忧宫(Sanssouci)似乎只是小木屋。17 世纪晚期,路易十四的英国和荷兰对手,把法国看作幅员辽阔、富有、强大、野心勃勃的陆地强权,时时在威胁整个欧洲的自由,有点像冷战期间美国人眼中的苏联。法国君主政体是欧洲国家建设进程中的急先锋,为建立现代中央行政国家打下基础。托克维尔在 19 世纪 40 年代写道,与他同代的法国人相信,他们的国家只是伴随法国大革命而出现的。如他所证明的,其基础却在两个世纪之前就打下了,法兰西王国的国王“越过大革命的深渊,与现代法国握手”。
同时,法国的国家大厦建造于腐烂和倾圮的地基。当 1715 年 9 月路易十四去世时,他的国家已彻底破产。皇家债务几达 20 亿里弗(livre),这还不包括 6 亿里弗的政府短期债券。法国的债权人已拥有直到 1721 年的未来税收,光是按期偿付连本带利的债务,便已超过可预期的未来税收。①这般险恶的财政并非新鲜事,路易十四的激进外交只是使之急剧恶化。法国国王为打造中央国家,持续一个多世纪,与地方有权有势者达成异常复杂的交易,后者以现金换取各式特权与豁免。国家逐渐蚕食所有百姓的自由,并以无法持久的方式将未来抵押给腐败的公职人员。它无法达到专制主义的更高阶段,像中国在数世纪之前所实现的。最后出于规范,它必须尊重它尝试掌控的社会阶层的利益,还必须尊重承继下来的法律。等到那些社会阶层被大革命的浪潮卷走,真正的现代国家才得以浮现。
在很多方面,法国君主政体的处境与当代发展中国家很相似,它们都把法治当作抵达目标路上的讨厌障碍。政府非常挥霍,将大笔资金投入战争,不愿花在补助金或社会福利上。由此而生的预算赤字必须找到资金,君主政体为此而四下寻觅。只要有逃之夭夭的可能,它都会牵强附会、扭曲、违反有关法律。但跑了和尚跑不了庙,最终,它还是要回到同一群债权人那里,去寻求新的资金。这种困境的唯一出路就是君主政体征用精英的财产,那也是大革命最终付诸实现的。但这超越了旧制度的想象力或能力,它因此发现自己陷入了永久的经济危机。
同时,政府寻求资金的对象,即法国社会,也无法反过来坚持负责制的基本原则。原因在于,不同经济阶层之间缺乏社会团结,或社会资本。贵族、资产阶级和农民,在更早历史时期曾有过团结,但现在彼此不抱同情。跟英国的情形不同,他们不相信自己是单一国家的一部分。这三个阶层内部又分化成自尊的等级,每一等级非常在乎自己的特权,以及相对于下一等级的优越地位,并不在乎政府掌控自己的阶层或国家。自由被当作特权,如托克维尔所说,在大革命的前夕,法国社会中“愿意为共同目标而一起奋斗的尚不满十人”。
在争夺主导地位的斗争中,中央国家和抵抗团体如果组织得不够严密,便出现弱的专制主义。法国的结局偏向于专制主义,但它非常脆弱,招架不住以人权为合法性基础的启蒙思想。
家族专制主义的起点
第一任波旁国王亨利四世在 1594 年加冕,其时,法国离统一国家或现代国家还很遥远。早期的法国国王以巴黎附近地区为权力基础,聚集数个公国,分别是勃艮第(Burgundy)、诺曼底(Normandy)、布列塔尼(Brittany)、纳瓦拉(Navarre)、朗格多克(Languedoc)。但在语言和习俗上,各区域之间仍有很大差异。王国一分为二,分别叫作财政区省(pays d’élections)和三级会议省(pays d’états)。前者是巴黎附近地区,构成国家的核心。后者是新近扩张的,处于疆土的边缘,使用不同的法律规则。此外,宗教改革又造成宗派分裂。天主教同盟和胡格诺派之间的宗教内战,要到原是新教徒的亨利四世皈依天主教,方告结束。他在 1598 年颁布南特敕令(Edict of Nantes),把天主教当作国教,但授予新教徒同等权利。
从波旁王朝到 1789 年大革命,法国的国家建设追随两条平行途径。第一条,法国国家愈益集权,从属单元的政治权利愈益缩小。这些从属单元早在封建时期便已存在,包括所有的公国,曾是地方政府的独立贵族,以及愈益纳入国家的保护和控制的市政厅、行会、教会乃至独立的私营商业组织。
第二条涉及集权的方式。不像早期的中国国家,也不像 18 世纪在勃兰登堡—普鲁士涌现的德国,法国的中央国家,并未建立在非人格化的任人唯贤的官僚机构基础上,因此也谈不上官僚职能专业化和教育。恰恰相反,它变成彻底的家族化。国家经常缺乏现金,急需收入,便把官位卖给最高投标者,从军事将领到财政部、征税官的公职,都可以拿钱来换。换言之,政府的私有化直达它的核心功能,公职都变成世袭的私人财产。②
如果以委托人和代理人的关系来理解廉政,代理人遵循委托人的指示必须得到鼓励。那么,法国政府所创造的制度绝对是一场梦魇。实际上,它给寻租和腐败披上了合法化和制度化的外衣,允许代理人在履行公职时谋取私利。事实上,租金一词(rente)就源自法国政府出售公职的实践,例如,出售征收特定税赋的权利,让买主获得长年累月的收入。③如果现代公共管理是公私分明,那么,法兰西王国代表了彻头彻尾的前现代制度。所以,法国国家只是现代和家族元素奇特而又不稳定的混合物。
中央行政国家和家族化公职的发展相互纠结,无法分开追踪它们的发展。法兰西王国的财政制度高度复杂,反映出它零敲碎打的发展过程。各种税项中最重要的是土地税(taille),直接征于农产品,由农民负担。还有人头税和一系列间接税,征于国内运输的酒和商品。国家垄断制造的食盐也须缴税(gabelle)。④后续的国王还征收其他税赋,包括人头税(人均税)和所得税(vingtième)。
直接财产税很难评估,因为没有制度来维持最新的人口普查,以及居民和资产的登记,像中国、奥斯曼和英国所做的那样。⑤富有家庭自然不愿诚实公开自己的资产,不然,他们的税赋就会上涨。⑥间接税的征收也很难,考虑到法国辽阔的疆土(如与英国相比)和分散的数千市场。17 世纪的法国经济尚未完全货币化,用来缴付现金税的硬币总是短缺。在这段时期,法国仍然是农业社会,那些在技术上容易收集的,如进口关税,尚没能提供实质性的收入。⑦
税赋制度的真正复杂性在于各种免税和特权。封建法国在中世纪晚期开发了两层会议的制度,一层是全国三级会议,另一层是一系列的地方或省级会议——又称为高等法院(sovereign courts, or parlements)——国王需要与之交换意见,以获得征收新税的许可。⑧为了鼓励各省加入法国的疆域,他授予省级会议特别的恩惠,承认地方精英的习俗和特权。税制因地区而有所不同,尤其是在财政区省和三级会议区省之间。贵族利用软弱的国王来为自己赢得各种豁免,从直接税到自产货物的消费税。这些免税和特权,开始自贵族向外扩散,抵达城市富有平民、皇家官员和各级地方官员等。赢不到免税的就是非精英者,即构成国家人口大多数的农民和工匠。⑨
公开出售公职的做法(venality,即捐官制或卖官鬻爵制)始于 16 世纪。法国为控制意大利,发动了与西班牙的持久战争,因此承担急需国家收入的压力。其时的国王光凭自己的收入尚不够支付战争费用,所以开始向意大利、瑞士、日耳曼南部新兴的金融中心举债。法国的信用从来不高,在 1557 年拒绝还债给“大借款”的银行家联盟后,更遭受极大的损害。它也拖欠为其打仗的外国雇佣军如瑞士人的薪金。在 1602 年,法国欠下 3,600 万里弗,债主是瑞士的州和市,以及指挥其军队的瑞士上校和上尉。法国政府一旦违约,瑞士雇佣军就停止参战。⑩
为了解决信用问题,国家的对策是通过一种租赁机制出售公职给私人。与普通放贷相比,租主享有该公职所控制的特定长期收入。他们至少在财政区省负责征收土地税和其他税项。由于税赋经过自己的手,他们得到取回本利的较大保障。内部财政(inside finance)的制度由此而生,国家财政的主要来源不再是私人银行家,而是已属国家机器一部分的富人。后者因自己的投资,而与国家沆瀣一气。
到头来,这些租金的信用也靠不住。政府很快将矛头指向租主,要求重新谈判相关条款。在亨利四世和财政部长叙利(Sully)治下,国家在 16 世纪早期想出一个新花样官职税(paulette):租主如果愿意付费,可将自己的公职转变成世袭财产,以传给后裔。⑪家族制的复辟可以从早期天主教会的改革中找到根源,那时教会为现代行政管理树立了一个先例,将圣俸从圣职中区分开来(参看第 18 章)。前者享有经济租金,它的传袭因神职人士的独身而受到限制;后者是功能性职位,并接受官僚等级制度的约束。但是,一旦非神职的平民进入国家官僚机构,因为没有圣俸或封建领地的许诺,便想方设法保住工作和照顾子女。法国政府也看到,让平民融入国家,变成了削弱古老贵族影响的有效措施。追求公职的最大客源是第三等级的资产阶级成员,他们希望购买公职来提高自己的身份。所以,全面家族化渗进了法国公共行政的核心。
官职税的采用并没终止国家筹款的诡计。国家将征收间接税的权利出售给包税商。后者在保证国家获得固定税金之后,得以保留额外的税收。国家也出售征收新税种附加税(droits aliénés)的权利,很快使传统土地税相形见绌。此外,国家增加出售公职的数量,以压抑现有公职的价格,从而稀释持有人的产权。对公职的如饥似渴,甚至令该制度的创建者感到惊讶。路易十四问他的财务总监蓬查特兰(Pontchartrain),他是如何找到购买公职的新人的。蓬查特兰回答:“陛下……国王一旦设定一份公职,上帝就会创造一名购买它的傻瓜。”⑫
该制度造成的低效和腐败非常可怕。财政部公职颇受欢迎,通常为私人金融家所购买,因为可以提前知道国家可能的招标,从而占据对付竞争对手的优势。财政部长定期主持汇票和其他财政记录的烧毁,以防秋后算账。⑬英国在发展公共财政和优化征税的高级理论,如亚当·斯密的《国富论》,而法国的征税却日益投机取巧、严重失调。⑭例如,法国各地的盐税高低不平,创造了人为的“盐税边界”,从而鼓励自低税地区朝高税地区的走私。⑮最重要的,法国财政制度特地鼓励寻租。富人不愿投资于私人经济中的产业,宁可购买不会创造财富只会重新分配的世袭公职。与其致力于技术革新,他们宁可挖空心思来与国家和税务制度斗智。这削弱了私人企业家的活力,使新兴的私人经济领域愈益依赖国家的援助。同时,英吉利海峡对面的私人市场却在蓬勃发展。
17 世纪晚期开发的法国财政制度相当落后,让穷人纳税,以支持有钱有势者。几乎每一个精英群体,从高级贵族、行会成员到资产阶级市镇,都为自己争取免税,把最沉重的税赋负担留给农民,这自然激起了一系列农民起义和反抗。为支持路易十四的战争而实施的增税,在 1661、1662、1663、1664、1665、1670、1673、1675 年都激起反抗。最后一次即是著名的法国红便帽起义。⑯它们一一遭受残酷的镇压。例如,1662 年的反税起义中,政府军带走五百八十四名俘虏,年过七十岁和不满二十岁的获得赦免,其余的都上了苦役船。⑰征税是为了支付军饷,但为了用武力执行征税任务,军队又必须自边境撤回,这不是在搬起石头砸自己的脚吗?它凸显了税收政策的根本教训:征税成本与百姓眼中征税当局的合法性,正好成反比。
总督和中央集权
17 世纪下半叶,在路易十三和首相黎塞留(Richelieu)、路易十四和马扎然(Mazarin)治下,法国财政危机以总督这一新建制为中央集权铺平道路。他们通常是年轻官员,前程全靠自己。如托克维尔所说,他们“并不是靠选举权、出身或买卖官职才获得手中权力”。重要的是,他们与地方精英或管理财政的鬻官等级制度全无瓜葛。总督通常是新近封爵的人,其直接下属即是平民。他们不像寻租者,巴黎的政府部门可随意予以辞退。中国为郡县配备官员,土耳其派人管理外省,现在法国发明了相同的制度。托克维尔继续说道:
然而,这些强势的官员在残余的古老封建贵族面前仍然黯然失色,仿佛消失于贵族所尚存的光芒之中……在政府内,贵族簇拥着国王,充实宫廷;他们统率舰队,指挥陆军。总而言之,贵族不仅是那个时代最令人瞩目的人物,连后代的眼光也常常停留在他们身上。若是有人提议任命大领主为总督,那便是对他的侮辱。最贫困潦倒的贵族,通常也会拒绝这样的职位。⑱
17 世纪中期之前,总督的派遣没有全盘计划,只是中央政府为应付特定麻烦而派出的。⑲渐渐地,他们愈益牵涉征税,尤其是传统上由地方官员监督的土地税。他们的篡权就是该世纪中期宪法危机的背景。
中央政府和地方参与者分享权力的斗争,主要涉及高等法院所发挥的作用。如前所述,法国有传统的两层会议制度。一层是省级会议,每省一个(其中最重要的是巴黎高等法院),另一层是全国三级会议。在中世纪晚期,法国国王定期召开全国三级会议来批准税赋,像英国议会一样。但没有它们,国王自己也能单独统治,这被视作专制权力的标志。从玛丽·德·美第奇摄政王的 1614 年,到大革命前夕的 1789 年,竟没召开过一次全国三级会议。代议制度在英国获得发展,在法国却没有。要弄清其中原委,必须了解高等法院为何在一国发展成为强大机构,在另外一国却没有。
代表地方精英利益的省级高等法院基本上是司法机构。跟全国三级会议不同,它们经常开会,可以成为对国王权力的制衡。国王如想颁布一项新税,就要来高等法院注册。高等法院通常举行公众讨论,遇上税务事项,会变得相当激烈。然后,高等法院可注册原封不动的法令,可修改,也可拒绝。不受欢迎的法令会在法庭上接受地方官员口头或书面的抗议。高等法院的权力很有限,因为国王可召开所谓的御前会议(lit de justice),将高等法院所拒绝的法令强行注册。⑳高等法院的抗议仅仅让国王蒙羞而已。
1648 年威斯特伐利亚和约(Peace of Westphalia)之后,该制度面临严重危机。其时,三十年战争的累计债款促使法国政府试图在和平时期继续战时的征税水平。巴黎高等法院的拒绝,最初导致马扎然打退堂鼓,从大多数的外省撤回总督。但高等法院领袖随后被捕,激起了所谓投石党(Fronde)的普遍叛乱。㉑从 1648 年到 1653 年,投石党运动分成两个阶段,代表了传统地方精英和贵族,对君主实施最终制裁,即武装叛乱。双方都有可能赢得内战,但到最后,政府政策激怒的各式社会参与者不能团结一致以取得军事胜利。
高等法院和贵族的失败,为法国政治制度的彻底集权铺平道路。17 世纪下半叶,路易十四和财务总监柯尔贝尔(Jean-Baptiste Colbert),故意将总督转化成国家工具,让皇家会议(Royal Council)赋予他们在全法国的统一权力。㉒他们被安插到每个省份,权力大为增加。他们开始招募和监督地方民兵,接管公共建设,负责公共秩序。救济穷人的义务,早已被封建贵族放弃,也变成由总督经手的中央政府的功能。㉓
国家建设过程中湮灭的自由,还包括城镇和市政的自治权。直到 17 世纪晚期,法国的城镇居民一直行使权利,以民主方式选出地方法官。他们维护自身权利,经常还获得国王的支持,作为削弱地方贵族的手段。㉔但到 1692 年,第一次废除选举,地方法官改成中央指派的总督。托克维尔对此作出评论:
值得历史大加蔑视的是,这场伟大的革命在并无任何政治目的的情况下完成了。路易十一之所以限制城市自由,是因为它的民主性质使他感到恐惧;路易十四之所以摧毁城市自由并非出于恐惧,真实情况是他把城市自由出售给所有能赎买的城市。其实他并不想废除城市自由,而是想以此为交易,即使他实际上废除了城市自由,那也绝非本意,而仅仅是基于财政目的的权宜之计。奇怪的是,这套把戏一成不变,竟然持续了八十年。㉕
托克维尔有一条非常有趣的评论。他所钦佩的新英格兰城镇是美国民主的基础,与中世纪的法国城镇一样,都源自相同的封建地方机构。到 18 世纪,两者却分道扬镳,原因在于法国中央政府的收买。㉖法国城镇政府开始受到寡头的控制,他们愈益通过买卖官职来获得公职,让自己出名。社区团结因而遭受破坏,除了掌控公职的精英,其他民众陷入冷漠。
政治集权的影响是非常深远的,建立了我们今天所知的更为划一的国家。1685 年撤销南特敕令,让天主教独霸一方,导致很多企业家和巧匠的新教徒移民到欧洲他处,甚至远赴北美和南非。中央政府现有更大权力,可以宣布新税,不用担心已被慑服的高等法院的反对,全国各地的税赋差异得以降低。投石党叛乱失败之后,贵族失去了其在农村的权力基础,反被召到宫廷。他们在那里直接为自己的补助金和免税进行游说,为觐见国王而忍受操纵。古老贵族争相出席路易十四的晨服仪式(levée),就是其中一例。贵族以真正的政治权力和财富作为代价,得以保留自己的社会地位。㉗仍然剩下的权力只是他们继续控制的领主法庭。我们在第 17 章中看到,此类法庭在英国逐渐纳入皇家的控制。所以,法国只在错误的地方获得统一:丧失地方上的政治自治,以致不能在社区问题上做出决定;保留了地方贵族掌控的不平等司法制度,以致人们更加不相信既有产权的公平。
中央集权的局限和改革的不可行
18 世纪早期,法国国家日益增强的权力践踏了个人权利,首当其冲的是产权。但它的做法,却是典型的欧洲方式,即通过操纵法律制度,而不是罔顾法律、纯用强力。要废除惯例的权利和约束,必须经过漫长的辩论,并依照封建法律秩序的规定,在政治上争个明白。因此,剥夺高等法院的权力,足足花费了将近一个世纪的时间。法国国王对反抗的农民非常残忍,对精英参与者却有不寻常的尊敬。在投石党叛乱遭受失败之后,两名带头造反的贵族蒂雷纳(Turenne)和孔代(Condé),要求并获得了路易十四的饶恕。如果这些人是中国贵族,他们和所有家人都会被处死。
路易十四死于 1715 年,身后的君主政体债台高筑。为了减少负债,国家诉诸类似保护费诈骗的伎俩。它掌控名叫司法堂(chambre de justice)的特别法庭,然后威胁要调查债权人的私人财务。几乎所有债权人或多或少都涉及腐败,便同意降低政府的欠债,以交换调查的取消。㉘用选择性的反腐调查来筹集收入,或胁迫政治对手,这种策略时至今日仍然流行。
新财政部长约翰·劳(John Law)上任后,法国尝试另一套应付债权人的办法。它创建国家银行,订出硬币换成钞票的固定比率,然后强迫百姓统统以硬币兑换钞票,如有不从,则以起诉、抄家、充公来威胁。过后,银行又毁约,让钞票在硬币的基础上一再贬值,实际上只想少付债务利息。约翰·劳宣称,个人手中的财产,只有用于国王认可的正当用途,方才真正属于个人,导致孟德斯鸠(Montesquieu)称他为“欧洲史上促进专制的最伟大人物之一”。但约翰·劳的制度最终证明无法实施,随后很快破产。㉙像近代的很多专政政体,法国君主政体发现,政治法令既不能建立投资者的信心,也无法取消经济的基本原理。
18 世纪时期,法国各式的社会和政治参与者,相互均势发生了重要改变。世界资本主义经济日益增长,提高了生产效率,导致物质财富和法国资产阶级的剧增。就重要性而言,这些经济变化却比不上同时发生的思想运动。关于人权和平等的启蒙思想,在欧洲迅速扩散,获得突如其来的胜利。18 世纪 80 年代重开三级会议,开会原因完全不同于先前:三级会议限制国王权力的权利,不再基于封建习俗的古老起源,而基于它们能代表享有平等权利的广泛公众。一般认为,法兰西王国的财政制度已变得非常可怕,既复杂又不公平。早先数代财政部长,使用各式花样来赖债和搜刮债权人,现在取而代之的是新见解:征税应该统一和公平,合法性来自法国人民推选的代表。
法国大革命和民主莅临的故事,大家都很熟悉,我不想在本书详尽叙述。我之所以提起,只是为了一个不同目的。18 世纪 70 年代和 80 年代的法国政治家,接受新思想的影响,尝试以和平改革的方式改造旧制度,但由于既得利益团体紧紧抓住政治权力不放,而屡屡受挫。
这样的努力有过两次。第一次始于路易十五和首相莫普(Maupeou)治下的 1771 年。莫普发起与高等法院的冲突,禁止他们彼此联系和举行罢工。对方拒绝听命后,莫普重组整个司法系统,并取消巴黎高等法院的大部分司法权。最重要的是,他废除司法等公职的出售,让由国王直接付薪的新法官取代寻租者。更为公平的新所得税也变成永久性的,所依据的是对资产更为严格和诚实的评估。政府由此向卖官鬻爵的整个制度发动正面进攻,所威胁的不但是捐官者的职位,而且是其家庭储蓄的投资。㉚
该行动引起极大反抗,反抗者既有捐官者的既得利益团体,也有新兴的民主公众,后者奋起支持寡头反抗专制权力的扩展。传统的家族精英,把自己对改革的反抗描绘成对独裁的抵制。颇不受欢迎的路易十五突然死于 1774 年,他的继承者路易十六(大革命期间被送上了断头台),最终被迫恢复高等法院所有的权利和特权。㉛
第二次是在杜尔哥(Anne-Robert-Jacques Turgot)担任财务总监的 18 世纪 70 年代。他是重农主义者,对政治改革不感兴趣,但深受自由经济思想的影响,希望使法国经济更趋合理化。在这点上,他很像现代发展中国家的财政部长。那些部长自己是技术专家,信奉新自由主义,在 20 世纪 80 年代晚期和 90 年代脱颖而出。杜尔哥废除了谷物的出口限制,以及旨在稳定面包价格的其他复杂规定。他进一步颁布法令,废除享有特权的行会,将劳役转换成地主的新税。所有这些,都可被视为现代化和理性的经济改革,在某种意义上,甚至是必需的。但它们遇上狂暴的抗议,不仅来自面临面包价格上涨的城镇穷人,还来自行会和其他依赖国家租金的既得利益团体。杜尔哥倒台,第二次努力终告结束。㉜
法兰西王国的政治制度无法自我革新。广大的寻租联合体获得权利,并在传统和法律中寻求保护,这就是国家权力的基础。他们的产权体现在公职中,但这是非理性和紊乱的,且多数又属不义之财。等到寻租者被非人格化和任人唯才的官僚所替代,现代法国方能涌现。如果政府正面攻击这些权利,就会使自己权力所依赖的法律制度变得非法。作为现代政治制度重要组件的法治,很早就在法国获得发展,远在负责制政治机构和资本主义之前。所以,它所保护的不是现代政治制度和自由市场经济,而是传统的社会特权和国家掌控的低效经济。即使等级制度高层,在思想上接受旧制度的破产和根本改革,他们也没有力量打破寻租联合体所建立的平衡。需要更为强大的力量,即制度外非精英团体的愤怒,借用革命来将之彻底摧毁。
抵抗法国专制主义的失败
如果说专制主义没在法国取得完全胜利,那么抵制它的社会团体,也没能向国家强加某种形式的政治负责制。事实上,后者的失败显然更为重要,源于他们未能团结一致、采取行动(参看图 2)。反抗场所应该是省级的高等法院和国家级的三级会议。这些法庭作出抗议、埋怨、辩论和抵抗,多次迫使法国君主政体撤回它们所反对的建议。但在革命前夕的三级会议之前,高等法院从没迫使君主政体接受自己高于行政机构的宪政原则。自然有人会问:这些封建时代遗留下的传统政治会议,为何没能参照英国的方式组织集体行动?这个问题并不局限于高等法院。在中世纪的英国和法国,城市也组织成自治的政治机构。为何前者最终发展成新英格兰城镇,后者却沦作被动的行政单位?
未以比较方式检视其他国家之前,我们尚不能回答这些问题。但我们可建议大致的分类,以缩小对有关原因的搜索。第一种解释,要在法国社会结构中寻找答案,如果不是更早,也要追溯到封建时代。政治学家托马斯·埃特曼认为,家族专制主义在法国、西班牙和意大利南部诺曼王国的兴起,肯定与罗马帝国崩溃之后那里自上而下的国家建设有关。卡洛林帝国之外的欧洲——英国、斯堪的纳维亚和部分东欧地区——平民和贵族之间存在更多的社会团结,并发展出了强大的基层政治机构,幸存至早期现代。在拉丁语的欧洲,这些地方机构的软弱,再加上中世纪以来的频繁战争,解释了应付专制主义的集体行动的缺席。日耳曼是卡洛林帝国的一部分,发展了非家族的专制主义。它不像西班牙和法国,没有那么早就陷入地理政治的激烈竞争。等到它真正面对军事威胁,可避免他人的错误,建立更为现代的官僚机构。㉝
托克维尔赞成的第二种解释,将法国失败归罪于并不遥远的近代。特别是他认为,法国贵族和平民之间缺少社会团结,是君主政体的故意挑拨所致。托克维尔解释说,欧洲各地的封建制度并没有特别悬殊的差异,庄园、城市、农庄都有类似的法律和社会团结。他的第二本著作《旧制度与大革命》,在第 9 章和第 10 章中提供了很多案例。地方上的法国领主和其平民属臣,每隔两星期出席领主法庭来裁判案件,就像英国的百户法庭。14 世纪的资产阶级在省级会议和三级会议中,都扮演积极的角色,只因遭到社会差别的排斥,才在后续世纪变得消极。“无代表即不纳税”的原则,在中世纪便已得到确立,不管是法国还是英国。㉞
对托克维尔来说,专制主义之下的法国社会不和,植根于家族制实践本身,并不植根于古代传统。“在人与人、阶层与阶层之间制造差别的所有方法,其中流毒最甚、最容易在不平等之上再添加孤立的,就是征税不公平。”麻烦始于 14 世纪下半叶:
我敢断言,自国王约翰被俘、查理六世疯癫而造成长期混乱、国民疲敝不堪之日起,国王便可以不经国民合作便确定普遍税则,而贵族只要自己享有免税权,就卑鄙地听凭国王向第三等级征税;从那一天起便种下了几乎全部弊病和祸害的根苗,它们折磨旧制度的余生,并最终导致它的暴毙。㉟
免税在所有特权中最受憎恨,随着税赋在 16 世纪和 17 世纪的稳定上涨而愈演愈烈。再加上卖官鬻爵,免税不只是某个社会阶层的特权,也变成个别家庭的特权。购买公职的个人,只要自己觉得安全,宁愿让同胞的权利受损。在英国,穷人享受免税特权。在法国,富人享受免税特权。
不平等的征税使人堕落,不管是贵族还是资产阶级。前者丧失他们的统治权,作为补偿,愈加死守世袭的社会地位。考虑到有那么多新近买爵的平民,旧贵族规定,很多公职需要候选人显示“四名祖先”(即四名祖父母)的贵族出身。暴发户自己挤入行列后,又尝试对后来者关上大门。资产阶级通过搬到城市和谋求公职,试图将自己与农民分开。他们原可投入企业活动的精力和雄心,现在转向公共权威所推崇的地位和安全。㊱
这还不是解释的终止。捐官和特权也存在于英国,但英国君主政体从没像法国那么有效地破坏议会团体的团结。托克维尔自己也承认,英国贵族从一开始起,与其说是世袭的种姓制度,倒不如说是真正的执政贵族(最佳者的统治)。才华超众的英国平民加入贵族阶层,比在其他欧洲社会更为容易,原因源自历史早期,现已模糊不清。我们再一次回到支撑龟的问题。很有可能,公职家族化本身也有赖一系列先在的社会条件,甚至被有意的政府政策所鼓励。
寻租的社会
法兰西王国就是今天所谓寻租社会的早期原型。在这样的社会中,精英花费所有时间来攫取公职,以保证自己的租金收入——在法国的例子中,那就是可以私用的长期固定收入的法律权利。
寻租联合体稳定吗?它持续几乎两个世纪,为法国作为主要大陆政权的崛起提供了政治基础。另一方面,我们知道冠冕堂皇的法国宫廷掩盖着严重的虚弱。最重要的是联合体之外的人们,都感受到愤怒和不公,这种感觉鲜明而强烈,最终在大革命中爆发出来。甚至联合体内的人,也不相信它的原则。如能彻底废除卖官鬻爵,君主政体会很高兴,曾在王国末期作出尝试。寻租者只顾自己,对他人存有很少同情。他们自己已深深陷入这个制度,所以无法容忍改革的想法。这是完美的集体行动难题:废除该制度,社会整体会受益匪浅;但制度参与者出于个人利益,便会阻止合作和变更。关于政治发展中法治的作用,法国的例子提供了教训。现代国家存在之前,法治便已出现于中世纪。它约束暴政,但也约束现代国家的建设。为了引进真正的现代社会,必须废除它所护卫的旧社会阶层和习俗。早期现代时期,对抗君主政体以捍卫自由,实际上是在保护传统的封建秩序和世袭的封建产权。而这封建产权,恰恰又与现代资本主义的经济秩序水火不容。政府觉得,它必须尊重传统精英的产权,既然不能直接征用,只好诉诸借贷和愈益离奇的财政花招。于是,家族统治如鱼得水。国家对法治的尊敬,反而帮助建立了高度不平等的社会,虽然尝试染指寡头精英的财富,但终告失败。所以,它只能在穷人和政治弱者身上筹集收入,从而加剧不平等,并为自己的灭亡铺平道路。 法国古老的家族制在革命中死去。不过,西班牙旧政权却创建了类似的制度,在 18 世纪躲过革命和改革,并将之输往拉丁美洲,后者不得不与这份遗产长期共处。
第 24 章 家族化跨越大西洋
拉丁美洲政府的特征未见于世界其他地区;早期现代的西班牙发展出与法国类似的家族专制主义;西班牙制度和其移植至新大陆殖民地
拉丁美洲大陆在地理、种族、文化和经济上具有极大的多样性,但各国又显示出共同特征,使拉丁美洲的政府模式,与东南亚、中东和非洲迥然不同。
到 21 世纪早期,拉丁美洲人口的大多数居住在世界银行标为“上中等收入”的国家。他们的年度人均收入在 4,000 到 12,000 美元之间,不但超过非洲的大部分国家,甚至超过快速增长的新兴国家,如印度和中国。①然而,经济增长趋于跳跃式,平均来看,仍远远低于 20 世纪中期以来的东亚国家。②第三波民主化以来,它在总体上成为世界上最民主的地区之一。随着民粹政府的兴起,例如在委内瑞拉,也出现了民主倒退。③
拉丁美洲在两个方面表现平平。第一是平等。该地区在收入和财富的不均上名列世界前茅。21 世纪的头十年,某些国家的不均水平略有下降,但仍相当顽固。④第二是法治。举行选举,使用民主负责制来摆脱不得人心的领袖,拉丁美洲国家做得都不错,但司法的日常工作却比较落后。这体现在治安不良、犯罪率居高不下、法庭程序堵塞、脆弱或无保障的产权、很多富人和强人的胡作非为。
这两个现象——不平等和脆弱的法治——互有关联。法治的保护在拉丁美洲通常只适用于极少数人,如大企业主管或工会成员。在秘鲁、玻利维亚和墨西哥,多达 60% 到 70% 的人口生存于所谓的非正式领域(informal sector)。这些人经常没有自己住家的房契,从事无照的商业,如果受雇,也不是工会成员,得不到正式的劳工保护。很多贫困的巴西人住在蔓延的贫民窟(favelas),政府当局袖手旁观,正义经常私下解决,有时还得靠犯罪集团。执法不公平更促进了经济不公平,穷人居住的世界基本上得不到法律保护。他们不愿投资于自己的家,因为没有明确的法律文件。他们身受犯罪之害时,也不愿信任警察。⑤
要发现不平等的来源很容易,其大部分都是承继下来的。很多古老精英的富有家庭是大地主,其祖先建立大庄园,又将之顺利传给后裔。很多拉丁美洲国家的财政制度,又使不平等得到进一步深化。经济合作与发展组织(Organization for Economic Cooperation and Development)的富裕国家,其财政制度主要用于从富人到穷人的再分配。它的实施可通过累进税制度(如美国),也可通过再分配政策,向低收入家庭提供资助和社会服务(如欧洲)。相比之下,拉丁美洲的财政制度只做很少的再分配,在某种情况下,再分配却给了相对优越的团体,像参加工会的公务员或大学生。正式领域的工人和各式精英,得以保住自己的福利和补助金。事实上,他们中的大多数在逃税方面相当成功。不像美国的累进个人所得税,拉丁美洲国家的税收很少来自个人。其富人擅长于隐藏自己的真正收入,或转移财产到海外,远离税务官的控制。这意味着,征税主要来自消费税、关税和增值税,落在穷人头上的便高得不成比例。
21 世纪初,拉丁美洲政府在管理宏观经济政策上大有长进,但这只是近况。其历史的大部分时期,拉丁美洲政府因预算赤字、公共部门大量举债、通货膨胀和国债违约而声名狼藉。⑥全洲范围的最后一次是在 20 世纪 80 年代初,墨西哥、巴西、阿根廷、秘鲁、玻利维亚和其他国家都宣告延期还债,通货膨胀随之猛升。阿根廷在 20 世纪 80 年代末经历了真正的恶性通货膨胀,年增长率超过 1,000%。它在 2001 年又一次面临财政崩溃和国债违约。
在政治上,拉丁美洲的统治也与众不同。如上所述,该地区近来有很好的民主记录。但在 20 世纪 60 年代和 70 年代,即古巴革命之后,该洲所有大国都屈服于军事独裁。虽然民主根源可追溯到 19 世纪早期第一个独立国家,但拉丁美洲没有一个政权其民主政府的历史始终不断。除了菲德尔·卡斯特罗的古巴,该地区的独裁政府没能建成可被称为极权主义的强国,也没能掌控足够的强制力,实施真正的社会革命,如剥夺富有精英的资产和收入。该地区的威权政府从没能采取极端措施(很幸运),像苏联或中国共产党政权下那样的集体化,或中国“文化大革命”那样的大规模死亡。做不到的还有“选举式威权”(electoral authoritarian)政权,如查韦斯的委内瑞拉,它们甚至无法控制政权本身的犯罪或腐败。⑦国家权力的伤害,大多落在非精英身上。如 20 世纪 80 年代,危地马拉政府发动可怕的剿反,以反对原住民族的游击队运动。富有的精英学会与非民主政府和平共处,避开国家权力的锋芒,经常获益于制度化的腐败。
如果这听起来亲切,那是因为这使人忆起法兰西王国的统治模式。在拉丁美洲,这些先例来自非常相似的家族政权,即早期现代的西班牙。跟法国相似,西班牙专制国家在 1492 年之后勉强拼凑而成。由于无止境的战争,西班牙君主政体永远处于破产之中。它试图通过借贷来弥补预算赤字,但很快在债权人面前丧失信用,最终诉诸像法国一样的各式伎俩来筹集资金,包括债务一再重整、货币贬值和出售公职。事实上,这个外强中干的国家为了搜寻现金,将愈来愈多的公职,包括大部分军队,都售给私人企业家。其结果是如出一辙的内部财政,私人成功地获取了国家创造的寻租权。贪污现象比比皆是,卖官鬻爵完全腐蚀了公私之分。
同时,托克维尔所叙述的法国因素,也在西班牙削弱对专制主义的抵抗。贵族、士绅和第三等级,本来应该团结起来抵抗王室权力,但却由于国家向个人提供参与分享租金的机会,而陷入四分五裂。中世纪时,西班牙议会(Cortes,像法国高等法院和英国议会)必须批准新税。但到后来,它中止了其制衡国家权力的功能。对公职和级别差异的耿耿于怀,又阻碍了西班牙社会采取集体行动。
这就是移植到新世界的政治制度,借助于新西班牙(墨西哥)总督辖区(viceroyalty)和秘鲁总督辖区。此外,它治下的社会制度比欧洲的更为不平等。就像收复失地运动(Reconquista)之后的西班牙,新大陆也是军事征服得来的。但不像前摩尔人领土,新大陆有大量原住民。16 世纪 40 年代,在玻利维亚的波托西(Potosí)和墨西哥的萨卡特卡斯(Zacatecas)发现重要银矿,由此开创了庞大的采矿帝国。欧洲统治者享用开矿租金,做工的都是沦为奴隶的原住民劳力。编年史家注意到,奔赴新大陆的西班牙人,不是去做工,而是去当主人:他们“全靠印第安人的劳动、手工和汗水”。⑧从一开始,西班牙美洲的经济道德就不同于定居新英格兰殖民地的农民小地主。如果美国政治制度都以黑奴历史悠久的南方各州为基础,其结果就是拉丁美洲的殖民政府。
破产的西班牙国家
随着斐迪南(Ferdinand)和伊莎贝拉(Isabella)在 1469 年的联姻,现代西班牙国家迅速出现于世界舞台。该联姻合并了阿拉贡王国和卡斯提尔王国,再加上阿拉贡属下的领土加泰罗尼亚(Catalonia)、那不勒斯、西西里岛。联袂后的君主政体在 1492 年征服摩尔人的最后堡垒格拉纳达(Granada)。同年哥伦布前往新大陆,为西班牙争得西印度群岛(the Indies)。他们的孙子查理五世添加了包括低地国家(Low Countries)和弗朗什-孔泰(Franche-Comté)的勃艮第,到 1519 年当选为神圣罗马皇帝,更把奥地利哈布斯堡王朝的土地纳入版图。
16 世纪 20 年代,查理五世控制当时世界上最大的帝国。帝国形成是通过王朝同盟,而不是征服,这一事实造就了财政上的捉襟见肘,从而对国家制度发展的性质发生决定性的影响。查理五世和儿子腓力二世(Philip Ⅱ)只有卡斯提尔一个安全的征税基地(包括珍贵的新大陆殖民地),不能向帝国其他地区抽取资源来应付开支。⑨尽管如此,哈布斯堡君主政体在半岛之外担起了昂贵的担子。其中之一就是在 16 世纪发起与法国的持久战争,为了控制意大利,尤其是米兰公国。另外的是与荷兰联合省长达八十年的战争。最后,还有在日耳曼土地上发生的毁灭性的三十年战争。它由于法国首相黎塞留支持新教徒,而演变成一场泛欧大战。这段时期的战争,因开发了星状要塞(trace italienne),而变得异常昂贵。这种要塞不易遭受围攻炮火的伤害,但城防工程因此而变得格外拖延和艰辛。⑩所有这些战争费用,卡斯提尔纳税人承担了其中的 80%。⑪
尽管有来自新大陆的贵金属,这些昂贵的外务负担几乎压垮了西班牙的财政制度。在 16 世纪和 17 世纪,政府费用始终数倍于美洲殖民地的汇款。金银进口,从 16 世纪 30 年代和 40 年代的每年 20 万至 30 万枚达克特,增至 16 世纪末最高的每年 220 万枚。但仍跟不上增长的债务,它在同期从 120 万涨至 600 万枚。⑫
16 世纪早期的西班牙国王宁愿借贷,也不愿增税,很快发现自己的信用不佳。在 16 世纪 20 年代,债务服务费用就超过税入的三分之一。到西法持久战争结束的 1560 年,它已超过税入的 100%。⑬西班牙国王募集不到足够的资金来应付赤字,只好在 1557、1560、1575、1596、1607、1627、1647、1652、1660 和 1662 年宣布破产。⑭这些破产并没赖掉债务,更像今天所谓的债务重整。国王以这些债务属于高利贷为由,宣布延期偿付短期和浮动的债务,然后再跟债权人开始拖延和不怀好意的谈判。债权人被迫将旧债务换成一纸新契(juro al quitar),有资格分享未来的税收,就像法国的租金。这种债券未标日期,可以转让,最初年息是 7%,但要面对利率和本金的任意调整。通过这种债券,西班牙君主政体得以染指卡斯提尔社会精英的储蓄——神职人员、贵族、士绅、官僚等。最强大的债权人往往能获得较好条款,或者不受延期偿付的限制,或者让较弱的债权人承受债务重整。维多利亚公司(Vitoria)无法收到政府付款时,便拒绝偿付自己的债权人,包括“修道士、修道院、救济院、寡妇孤儿、其他非商人”。⑮政府发现,在政治上更难向这些精英直接征税,宁可选择不断赖账。这个传统也传至拉丁美洲的当代政府,如阿根廷。它在 2001 年的经济危机后,强迫外国投资者以及国内的养老金者和储户大量放弃手中的国债。
无代表仍纳税
当时很多欧洲人,尤其是受到西班牙威胁的英国人,对西班牙国王所谓的专制权力心存敬畏,相信他具有“像土耳其一样”的征税权和特权。但西班牙政权的财政基础却非常不稳定,国王对自己属下精英的权威也受法律和习俗的限制。西班牙的专制主义太弱,不敢像中国和俄罗斯那样向自己的精英发起正面进攻。它也无法像英国所做的那样开发基于情愿的合法征税制度。
像其他欧洲国家,聚集成西班牙的各王国都有称作议会的中世纪机构。莱昂王国(Kingdom of León)的议会是欧洲最古者之一,阿拉贡王国的议会是组织最好者之一,非常强势。⑯兼并莱昂的卡斯提尔王国议会,与英国议会或法国三级会议相比,其代表性少而限制多。它通常并不邀请作为集团的神职人员或贵族跟平民坐在一起开会。在 14 世纪,召集到议会的有一百座城镇的代表(procuradores),到 15 世纪,该数字跌至来自十八个城市的各两名代表。这三十六个人声称可代表全西班牙讲话,但实际上只是治内主要地区的寡头代表。⑰
议会的传统权力也受到限制。它没有立法权,因为已经留给国王。腓力二世在 1567 年颁布的新法典(Nueva Recopilacion)说,“一定要召集议会,征得代表的首肯,方能在整个王国征收税赋、捐献和其他税项”。但这指的是新设的额外税。像消费税(alcabala)、关税(regalias)、盐税及矿物开采税(quintos)的既存税,则不需要获得批准。国王也宣称,如果需求“合理”,议会无权拒绝。什么是合理,全凭国王说了算。
国王和议会的相对权力不是凭空而来的,而是政治斗争的结果。中央政府将消费税包给包税商,但遭到各城市的反对,后者宁要由自己负责收集和分配的人头税(encabezamiento)。人头税当年是伊莎贝拉批准的,1519 年被查理五世废除,从而激发所谓的公社叛乱(comuneros)。查理五世在议会安插自己心腹,不顾反对,强行通过新税。反对原因在于他被视为外国人(出生于佛兰德斯),向卡斯提尔征收的税,又用于不涉及本地利益的外国战争。卡斯提尔所有的城镇都奋起反抗,组织民兵,并要求另组民选议会,拥戴胡安娜女王(Queen Joanna)当政。要不是公社叛乱进而反对贵族,查理五世很可能丢失对王国的控制。贵族转而向国王靠拢,查理五世最终得以重建军事控制。⑱
公社叛乱的结果,在某种意义上,很像一百三十年后法国的投石党叛乱。国王以决定性的军事胜利宣称他对城市的权威。由民选的独立议会充任西班牙的自由保护人,这种想法彻底寿终正寝。同时,国王意识到他需要化解不满,遂逐一收买潜在的对手。当初激发叛乱就是因为人头税的废除,他现在予以恢复,还将服务税(servicios)和普遍税(millones)的新税留在地方当局手中。他们多半是家族官僚,帮国王征税,自己可保留一部分。⑲议会后来重开时,只提供咨询,再也没有要求或获得征税的权力。但他们的偏袒还是会影响公共财政,因为他们不愿支付财产税,所以新税都是影响穷人甚巨的商业税,从而阻碍西班牙的经济增长。
西班牙国家的家族化始于 16 世纪 60 年代,在腓力四世(1621—1665)治下到达顶峰。跟法国一样,驱动这一进程的是西班牙的持久战争和无止境赤字。西班牙第一次破产是在 1557 年,国王要他的朋友和侍臣鲁伊·戈麦斯(Ruy Gómez)去兜售市政公职,多多益善。⑳跟法国不同,西班牙的卖官鬻爵最初只是城市和地区的。该措施受到广泛谴责,大家知道售出的公职不能提供足够的回报,除非走歪门邪道。㉑尽管如此,财政困境促使国家出售更多公职。到了 1650 年,据估计政府共创造三万名捐官,按人均来算是同期法国的两倍。㉒此外,卡斯提尔领土的 30% 回归领主法庭的管辖,不是为了政治目的,而是因为君主政体急需现款。各城镇的全部权力,包括征税权和司法权,都出售给私人。在某个意义上,西班牙的国家建设开了倒车,由于财政上的短见,中央政府失去对大部分领土的控制。
家族制也影响军事组织。西班牙经历很多世纪,方从摩尔人的手中获得解放。卡斯提尔王国和阿拉贡王国联姻合并时,军队组成所谓的步兵方阵,配备长矛,以后又改成火绳枪(arquebus,编按:中国称鸟铳或鸟枪)。㉓如此训练和装备的西班牙军人,在科尔特斯(Cortés)和皮萨罗(Pizarro)的率领下,战胜了新大陆的本土帝国。他们也奔赴西班牙帝国的其他地区驻防,尤其是意大利北部的基地,从那里可经过所谓的西班牙路(Spanish Road)直达低地国家。㉔卡斯提尔士兵参与了 1533 年反对奥斯曼帝国的维也纳防御战。西班牙水兵也以少量舰队参与 1535 年进攻突尼斯(Tunis)、1538 年试图攻占阿尔及尔(Algiers)、1571 年重大的勒班陀战役(Battle of Lepanto)。到了 17 世纪,募集陆海军的任务越来越多地交托给自资招募军人的私人和装备自己舰船的沿海城镇。向军队供应必需品的后勤基础,又受控于热那亚(Genoa)的金融家。这意味着,到 17 世纪中期,西班牙君主政体对属下的武装力量只行使很有限的控制。㉕
像其他西欧国家,法治扮演了重要角色,限制了西班牙国王在产权和公众自由方面的权力。跟北欧不同,罗马法的传统在西班牙没有完全消失。《查士丁尼法典》重现于 11 世纪之后,西班牙发展了颇为强大的民法传统,民法被视作神法和自然法的成文化。国王可颁布制定法,但新法典讲得很清楚,必须遵循既存的法律先例,与之相悖的皇家法令则没有效用。天主教会仍是教法的监护人,并经常向皇家特权挑战。与习惯权利和特权相抵触的皇家命令常常受到抵制,此举被称作“服从但不执行”(Obédezcase, pero no se cumpla)。赴新大陆的征服者(conquistadore),如果从总督辖区接到自己不喜欢的命令,经常援引此理。个人如不同意收到的皇家命令,有权向皇家会议提出申诉。后者像英国的对应物,享有西班牙的最高司法权。根据历史学家汤普森(I. A. A. Thompson),卡斯提尔的皇家会议信奉条文主义(legalism)和正当程序,反对随心所欲。它还主张相对于行政模式的司法模式,积极抵制非正常程序,始终保障既定的权利和契约义务。㉖
该法律传统的影响,体现在西班牙国王如何处置国内敌人和百姓产权。在西班牙,找不到秦始皇或伊凡雷帝(Ivan the Terrible)那样的帝王,他们会任意处决自己宫廷的成员,以至灭族。像同期的法国国王,西班牙君主在搜索财源中不断侵犯国人产权,但仍在现有法律的框架中运行。他们没有任意征用资产,只是重新谈判利率和本金的偿还表;不愿增税以造成对抗,只是使货币贬值,承受较高的通货膨胀。滥发货币的通货膨胀实际上也是一种税赋,但无须通过立法,对普通百姓的伤害超过精英,后者拥有的大多是实物资产,而不是货币资产。
制度移植到新大陆
与长期定居、拥有古代习俗的社会相比,征服社会为制度的发展和改革提供了不同的机会。征服社会可实施当代企业所戏称的“未开发地区的发展”——不受既得利益团体和习惯行为的妨碍,彻底重建制度。奥斯曼帝国在封地上安顿骑士,使之成为仅一代的贵族,因为土地是不久前抢来的。一点也不令人惊讶,西班牙征服新大陆时,随身带来了现成制度。与欧洲相比,他们面对更少既得利益者的遏制,以及不同的经济机会和自然资源。如果拉丁美洲的统治类似于西班牙王国的统治,制度移植却不一定直截了当,或刻不容缓。
收复失地运动的最后战役之后,接踵而来的就是西班牙征服美洲:哥伦布(Christopher Columbus)目睹斐迪南和伊莎贝拉在格拉纳达凯旋入城;科尔特斯的叔叔和父亲参与反对摩尔人的战役。科尔特斯在与阿兹特克人(Aztec)打仗时,好像仍在与摩尔人作战,运用分而治之的类似策略。㉗
很多有关定居、殖民和政治制度的技术,直接搬自西班牙南部的殖民经验。事实上,征服者习惯把美洲本土庙宇称作“清真寺”。
这些早期探险受到西班牙国王的资助,但主要依靠组织探险的私人企业家的能量。一边是身处新大陆的个人,另一边是尝试严控殖民地的马德里政府,两者之间的互动造就了拉丁美洲的制度发展。金银开采权利特别重要,因此颁给私人的土地不包括地下权益,全部留给国家。赴秘鲁和墨西哥的大部分移民,并不涉及金银的开采。更确切地说,他们只想充任土地和由此而生的农业资源的主人。与西班牙南部相比,他们面对全新环境,所征服的土地住有密集人口,适合不同模式的开发。
为了奖励和控制征服者,西班牙当局发明了托管权(encomienda)制度,所赠予的不是土地,而是原住居民。如奥斯曼帝国的封地,国王的意图是防止既得利益的地方贵族兴起。托管权的赠予是有条件的,不得遗传。㉘科尔特斯征服了阿兹特克首都特诺奇提特兰(Tenochtitlán),其幸存属下中大约有 40% 获得托管权,相当多的皮萨罗追随者在秘鲁也获得托管权。从技术角度看,托管权并不将原住民当作奴隶,但要求他们提供劳力,以换取监护者给他们的基督教教育和善待。西班牙国王以家长姿态,担忧新主人虐待原住民工人,也担忧天花和其他极易为印象第安人感染的疾病造成原住民人口急剧下降。所以,基于种族的主奴等级关系成为早期拉丁美洲制度的组成部分。
为统治美洲殖民地,西班牙迅速建立了相对有效的现代行政机构。西班牙新大陆帝国的合法性来自教皇亚历山大六世 1493 年的诏书,它将西印度群岛(地理范围不明)永远赐给卡斯提尔和莱昂的国王。权力属于西班牙国王和马德里的西印度群岛理事会,再传至设立于墨西哥和秘鲁的总督辖区。用于新大陆的法律是卡斯提尔的,与帝国其他地区毫无关联,尽管很多西班牙征服者和新移民出生于他处。科尔特斯在 1519 年开始对墨西哥的征服,下一年就发生重大的公社叛乱。由于这场叛乱,移植到新大陆的政治制度不包括强大的议会,或其他类型的代议制度。政治独立的唯一努力来自皮萨罗的兄弟贡萨罗(Gonzalo),他尝试成为独立的秘鲁国王,在 1548 年被皇家军队打败并处决。自那以后,中央权力再也没有受到新大陆西班牙人的挑战,直到 19 世纪早期的独立战争。
西班牙当局移植罗马法律制度,在十处建立高级法庭(audiencia),包括圣多明各、墨西哥、秘鲁、危地马拉、波哥大。派去帮助治理殖民地的行政人士中,有很多是具丰富民法经验的律师和法官。行政人员不得与本地女子结婚,或在领地上建立家庭联系,很像中国的县令或奥斯曼帝国的桑贾克贝伊。历史学家约翰·赫克斯泰布尔·艾略特(J. H. Elliott)在评论殖民地政府时写道:“如果现代国家中的‘现代性’,指的是将中央权力的指令传达到遥远地区的机构,那么西班牙美洲殖民政府要比西班牙政府,甚至其他任何早期现代的欧洲国家,更为‘现代’。”㉙在这一方面,它与英国君主政体对北美殖民地的放任态度,形成鲜明的对比。
大庄园的铁律
1570 年在新大陆的西班牙行政机构,似乎比同时代的欧洲制度更为现代,但好景不长。西班牙政治制度的家族化要到 17 世纪才加大油门,卖官鬻爵之类的制度移植到新大陆也属无可避免。推动这个过程的基本动力,来自殖民地实际参与者的倡议。他们试图增加自己的租金和特权,而马德里的中央政府太软弱、太遥远,无法予以制止。
大地产或大庄园的铁律——富人将变得更富,除非遭到国家的遏制——既适用于像中国和土耳其那样的农业社会,也适用于拉丁美洲。移民阶层强烈抵制托管权仅维持一代的规定。一点也不奇怪,他们要求将自己的权利传给孩子,便在 16 世纪 40 年代公开违抗托管权自动回归国王的法律。拥有原住民的劳力,使部分托管权主人发财致富,并开始购买大片土地。不像托管权,土地可以遗传。到 16 世纪晚期,美洲面对本土居民濒临灭绝的危机。墨西哥的人口从 2000 万跌至 160 万。㉚这意味着许多人口稀少的土地突然进入市场。
新兴的克里奥尔(creole,编按:指生于美洲的西班牙白种人)精英大多住在城镇,雇用劳力开发土地,自己只是缺席地主。拉丁美洲惯例的土地所有制,与其他部落社会相比,基本上没有很大差异。产权共有,并联系着扩展的血缘团体。剩下的印第安人,要么受骗售出自己的土地,要么被人赶走。共有土地变成私人地产,由于玉米和木薯等本地作物被欧洲经济作物所取代,周遭环境大变。很多农地转换成养牛牧场,对土壤肥力造成极大损害。马德里政府承诺保护原住民地主的权利,但天高皇帝远,无法控制实际局势。而地方上的西班牙当局往往与新兴的地主阶层狼狈为奸,帮助后者逃避有关规则。这就是拉丁美洲大庄园(hacienda)的起源,在后续年代里,成为不平等和持久冲突的根源。㉛
少量精英却拥有大片土地,在西班牙长子继承权(mayorazgo)实践中找到支持。它防止土地的分割出售和大庄园的瓦解。17 世纪见证了富人的大肆兼并,甚至是整座村庄和城镇。他们再借用长子继承权,以防遗产分配造成土地流失。长子继承权也已移植至新大陆。西班牙当局试图限制长子继承权的牌照,所依据的道理与他们要求收回托管权一样。地方上的克里奥尔或移民群体,转而使用改进继承权(mejora)。父母在遗产分配上可作偏袒,目的仍是维持宗族的实力和地位。㉜
强大的地主阶层出现,但无法成为凝聚的政治参与者。像法兰西王国,税务制度帮助将个别移民与国家绑在一起,破坏了他们可能建起的与非欧洲同胞的团结。构成早期移民浪潮的有大批单身男子,结果与本土女子要么结婚,要么生孩子,造就了麦士蒂索混血阶层(mestizo)。愈来愈多的黑奴运来新大陆,与白人一起生下的后代叫穆拉托(mulatto),成为又一单独阶层。区别于这两个阶层,西班牙移民的后裔克里奥尔可以享受免税。这种待遇,如在西班牙,只属于贵族和士绅(hidalgo)。就像在北美,身为白人就能获得地位,截然不同于恭恭敬敬的印第安人和黑人。㉝
考虑到国王在马德里的财政拮据,卖官鬻爵的欧洲制度最终越过大西洋也许是不可避免的。西班牙美洲的财政管理,在 16 世纪的大部都还不错。殖民地毕竟是贵金属的主要来源,再逐渐改为农产品。到世纪末,矿产开始下跌。随着三十年战争的进行,西班牙国王对税收的需求又有增加。君主政体防止新大陆出现贵族阶层的努力,因此而销声匿迹。艾略特如此描述这个转变:
城市的主要家庭借助与皇家管理机构的特殊关系,聚积资源,按自己需求建立继承权,巩固对城镇和内地的掌控。他们还利用国王日益恶化的财政困境,趁机购买公职。市政会职位(regimiento)的私人交易由来已久,从 1591 年起,更变成公开出售。从 1559 年起,公证官的职位上市。到 1606 年,几乎所有地方公职都跟进买卖。腓力二世和腓力三世反对出售财政部的公职,但到 1633 年,腓力四世开始放开买卖。最终,到 17 世纪下半叶,甚至最高级职位也上了市场。从 1687 年起,就系统性地出售高级法庭的职位。㉞
像法国和西班牙,对商人阶层来说,购买公职成为提高社会地位的途径。他们现在把自己当作绅士(caballero),将来再传给孩子。古老家庭更可购买西班牙贵族的爵位,以保护他们相对的优越地位。17 世纪的西班牙君主敞开大门,允许数百名克里奥尔进入颇有声望的西班牙军事修道会(Military order),分封其余的为侯爵和伯爵。
到 18 世纪,平等和人权的原则开始向新大陆殖民地渗透,但西班牙政治制度和社会制度已在拉丁美洲获得再生。讽刺的是,家族制度的移植违背了马德里殖民当局的初衷。在 16 世纪的大部分时间,他们尝试在殖民地建立更为现代的非人格化政治秩序,但这些计划均因国王日益恶化的财政而搁浅,使他们难以实施更为强硬的遥控。伊比利亚半岛上出现的公私不分,也在美洲发生。
在法国,寻租者和捐官者攫取国家,破坏国家权力,最终造成法国大革命的社会爆炸。在西班牙,相同的政治演变造成国力的长期衰退。但类似的政治革命,从没光顾西班牙的母国或殖民地。19 世纪早期的独立战争推崇法国大革命和美国革命的自由和平等,但其领袖是克里奥尔的精英——像西蒙·玻利瓦尔(Simón Bolívar)——他们曾深深陷入旧政权的家族政制。
法国大革命得以在公共利益和私人利益之间重新划定明确界限。它没收所有捐官者的世袭财产和特权,谁反抗就砍谁的头。新式的政治制度,其公职的招聘基于非人格化的选贤与能——中国人在将近两千年之前就已发明的——又由马背上的拿破仑带往欧洲其他国家。1806 年,他在耶拿和奥尔斯塔特(Jena-Auerstadt)两次击败普鲁士的家族化军队,从而说服新一代的改革家,像冯·施泰因男爵(Baron vom Stein)和卡尔·奥古斯特·冯·哈登贝格(Karl August von Hardenberg),普鲁士国家必须以现代原则进行重建。㉟19 世纪的德国官僚机构,成为韦伯现代合理政府的模型。它并不来自家族化官僚,而是与传统的刻意分手。㊱
在拉丁美洲,独立成功之前从没发生社会革命,家族制仍然嵌入很多独立后的政权。虽然出售公职和贵族封号的做法遭到废除,正式的民主制度获得建立,但旧心态依旧长存。19 世纪拉丁美洲的新国家中,很少强大到能直面自己的精英,或加以征税,或加以抑制。那些精英渗透和控制国家本身,并找到空隙,将自己社会和政治的特权传给孩子。直到 20 世纪晚期,西班牙旧政权的财政恶习,像持续赤字、过分借贷、债务重新谈判、隐性征税的通货膨胀,仍在阿根廷、墨西哥、秘鲁、玻利维亚等国徘徊。正式的民主和宪政,并不基于社会各阶层的对抗和妥协,而是精英自上而下所施与的,如果不再符合自身利益,又可收回。这引发高度不平等和两极分化的社会在 20 世纪的涌现,并酿造了真正的社会革命力量——体现于墨西哥和古巴的革命。过去一世纪中,拉丁美洲国家定期遭遇骚乱,要求对整个社会契约进行重新谈判。
近来出现很多新兴的社会参与者,譬如工会、有密切国际关系的商业团体、城市知识分子、试图要回殖民者所夺走的地位和权力的原住民团体。拉丁美洲政治制度的对策,不管是民主的还是威权的,都趋于让他们一步步参与国家,从而收买他们,而不是政治权力真正的重新调整。例如在阿根廷,20 世纪初的前数十年,工人阶级的兴起遇到传统地主精英的顽强抵抗。在欧洲,工人阶级加入广泛组合的社会民主党,提倡再分配政策,为现代福利国家打下基础。相比之下,代表阿根廷工人阶级的却是军事领袖胡安·庇隆(Juan Perón)。他的阿根廷正义党(Partido Justicialista),向拥护者网络提供选择性的好处。阿根廷在民粹狂热和军事独裁之间左右摇摆,并没开发出真正欧洲风格的福利国家。革命制度党(Partido Revolucionario Institucional)治下的墨西哥也有类似情形,特别优惠只给选出的组织良好的拥护者。墨西哥比阿根廷更稳定,但同样无法解决社会隔绝和贫穷的难题。所以,西班牙旧政权的家族遗产仍在 21 世纪存活。
第 25 章 易北河以东
匈牙利成为失败负责制的另一选择;西方废除的农奴制却在东欧冒头;宪政主义和贵族统治出现于匈牙利;自由如要兴盛,既要有强大中央国家,又要有对它的制约
早期现代的法国和西班牙是弱的专制主义和失败负责制的案例。形成于 16 世纪和 17 世纪的国家是专制的,因为它的君主政体集中权力,无须以正式方式向议会或其他代议机构负责。其他政治和社会的参与者,如高等法院和西班牙议会,公社叛乱者和投石党人,反对国家集权,最终都被一一击败。他们失败的方式凸现了专制权力的基本弱点。国家向精英参与者提供一部分国家职能,将他们逐一收买,既削弱了他们集体行动的能力,也限制了可在他们身上行使的权威。他们的财产和特权,虽然经常受到挑战和侵蚀,但基本上完整无缺。
相比之下,匈牙利和俄罗斯提供了两条另类发展路径,它们彼此之间不同,又有别于法国和西班牙的模式。这四个案例最后都以政治负责制的缺席而告终。在匈牙利,专制努力最初是失败的,因为强大和组织良好的贵族阶层,可以向国王权力施加宪法的限制。跟英国议会一样,匈牙利议会也迫使匈牙利国王向自己负责。但他们对负责制的追求,并不代表全体国民,只代表狭隘的寡头阶层;他们只想使用这份自由,以进一步榨取自己属下的农民,又避免向中央国家缴纳重税。其结果是愈益恶劣的农奴制得到扩张,国家趋于孱弱,最终不能抵抗土耳其。换言之,一个阶层的自由导致了其余阶层的不自由,还导致国土被强大邻国宰割。
我们花时间来考虑匈牙利的例子,只想显示,对中央政府权力的宪法限制,并不一定能建成政治负责制。匈牙利贵族阶层所追求的,是更加彻底地剥削农民的“自由”,强大中央国家的缺席让他们得逞。大家都理解出自中央专政之手的中国式暴政,但暴政也可来自分散的寡头统治。真正的自由倾向于在社会精英参与者的均势中出现,匈牙利从没能做到这一点。
主人和奴隶
欧洲历史中重大谜团之一是早期现代之初,即 16 世纪和 17 世纪,主子和奴隶的关系在东西欧得到截然不同的发展。易北河以西的地区——西部日耳曼国家、低地国家、法国、英国和意大利——中世纪期间强加于农民的农奴制逐渐取消。奴隶制从未在西班牙、瑞典和挪威出现。相比之下,易北河以东的地区——波希米亚(Bohemia)、西里西亚(Silesia)、匈牙利、普鲁士、利沃尼亚(Livonia)、波兰、立陶宛和俄罗斯——先前自由的农民却在历史同期逐渐沦为农奴。①
跟封建制一样,农奴制有繁多定义。历史学家杰罗姆·布鲁姆认为,“如果一个农民受领主愿望的束缚,相互之间的关系使他低人一等,并在社会中无能为力;这种情形又被认作是领地上法律和社会结构的根本,而不是领主与他的契约或协议的结果;这样农民就是不自由的”。对农民享有司法权的是领主,而不是国家。他们的关系可由详细的惯例规则所定位,但领主可以修改规则,使之更加不利于农民。农奴仅保留少许的法律权利,不同于奴隶,但实际差别并不大。②
从 12 世纪以来,西欧农奴在不同时期和不同程度上赢得自由。他们通常先升为领主土地上的佃户,土地使用权可能限于自己的一生,也可能传给孩子。有些土地受到限制(mainmortable),只能传给与自己同住的孩子,否则就要归回领主。在 18 世纪,废除该限制成为自由改革家的重要目标之一。在其他案例中,农民直接升为地主,享有随意买卖和赠与土地的全部权利。法国大革命的前夕,法国农民已拥有土地的 50%,超过贵族的两倍。③托克维尔指出,那时的领主早已停止参与对农民的统治。这就是残留的收费权利,或迫使农民使用领主的磨坊或酒坊,受到如此强烈憎恨的原因。④
在东欧发生的情形恰恰相反。中世纪时期,与西方相比,那里反而有相当充分的自由。多半因为它仍属人口稀少的边境地带,来自西欧和欧亚大陆的殖民者,可遵循自己的法律。从 15 世纪开始,整个东欧建立新规则,限制农民的迁徙。农民不得离开他耕耘的土地,否则就要面对大笔罚款的威胁。帮助潜逃农奴要受沉重处罚,城镇收容农民的能力大受限制,以防止他们逃避庄园上的义务。
农民损失最大自由的是俄罗斯。回溯到 12 世纪的基辅罗斯(Kievan Rus),其时已有奴隶和农奴。随着 15 世纪莫斯科国家的兴起,农民的义务持续上升,活动自由也在逐渐减少,直到每年仅得一次假(前提是债务已经还清),在圣乔治节(St George’s day)的前后。到了下一世纪,连这唯一年假也被取消。⑤俄罗斯领主对农奴的权利稳步加强,直到 18 世纪末。其时,人权原则正在整个西方传播。农奴永久绑在主人身上,没有活动权利。事实上,主人可随意调遣农奴,从一处地产迁到另一处,甚至将农奴放逐到西伯利亚,之后又任意召回。俄国统治阶层开始以手下农奴的数量来评估自己的地位。俄罗斯的贵族高层富得惊人:伯爵尼古拉·谢列梅捷沃(N. P. Sheremetov)拥有 185,610 名农奴;到他儿子手上,这个数字增至 30 多万。18 世纪末,伯爵沃龙佐夫(Vorontsov)拥有 54,703 名农奴;到 19 世纪中期农奴制废除之前,他的继承者光是男奴就有 37,702 名。⑥
农奴制在东西欧为何有如此迥异的发展?答案在于经济、人口和政治因素的总汇,使农奴制在西方难以维持,在东方却盈利丰厚。
西欧人口密集,在 1300 年是东欧的三倍。随着始于 11 世纪的经济繁荣,众多人口变成城市居民。这些城市从意大利的北部辐射至佛兰德斯,其存在首先是政治软弱的产物,再就是国王发现,保护城市的独立可以挖对手大领主的墙脚。城市也受到古老封建权利的保护,罗马时代的城市传统并未消失。由于受到如此庇护,城市发展成为独立的社区,通过贸易增长来开拓自己的资源,独立于庄园经济。⑦自由城市的存在,又使农奴制愈加难以维持。它们好像是国内的边境线,农奴可以逃到那里来赢得自由,因此有中世纪的说法,“城市空气使你自由”(Stadtluft macht frei)。⑧相比之下,人口相对稀少的东欧城市更为小型,跟中国和中东类似,主要充任现有政治权力的行政中心。
14 世纪的灾难性人口下降,更促使西欧趋向自由和东欧趋向非自由。重复发生的瘟疫和饥荒对西欧的打击,比对东欧更为严峻,爆发时间也更早。经济增长在 15 世纪恢复,西欧看到城镇的再生。它们提供避难所和经济机会,防止贵族进一步榨取手下的农民。事实上,为了挽留农民继续耕耘,领主必须提供更多自由,从而开启了现代的劳动市场。中央君主政体发现,保护城镇的权利可以削弱贵族对手。日益增加的需求必须依赖来自东欧和中欧的进口,包括食物和贵金属。但在易北河的东面,软弱的独立城市和国王,允许贵族依靠农民劳力来开发农产品的出口。如历史学家杰诺·苏克斯所说:“从长远看,易北河对岸的地区为西方复苏作出了贡献……‘第二次农奴制’的立法凶兆,以可怕的同步出现于勃兰登堡(1494)、波兰(1496)、波希米亚(1497)、匈牙利(1492、1498)、俄罗斯(1497)。”⑨
这是对东西欧农民权利不同模式的最明显解释。在西方,愈益强大的国王支持城市,其存在可以抵消贵族权力。在法国和西班牙,国王最终在长期斗争中获胜。与领主有委屈或冲突的农民和其他参与者,从精英的竞争中获得更多机会。在东欧,城市和君主权力都很弱,让贵族阶层自由支配属下的农民。这样的模式出现于匈牙利和波兰,国王由贵族阶层选出。东欧两个地区有强大国家:15 世纪之后的俄罗斯和 18 世纪之后的勃兰登堡—普鲁士。然而,在这两个案例中,国家都没有代表平民来反对贵族,反而联合贵族来反对农民和资产阶级,再招聘贵族服务以增加自己的权力。
后来,农民在大规模行动中获得解放,例如沙皇亚历山大二世 1861 年的解放宣言。但非精英的真正自由——不仅农民还有城市中的工匠和资产阶级——还需依赖现有精英参与者的僵局或均势。非精英团体在两种情况下都受到压榨:第一,分散寡头变得太强大,那是匈牙利和波兰的情形;第二,中央政府变得太强大,那是俄罗斯的情形。
宪政主义及其在匈牙利的衰落
今日匈牙利只是中世纪幅员辽阔王国的缩影,它曾在不同时期囊括今日奥地利、波兰、罗马尼亚、克罗地亚、波斯尼亚、斯洛文尼亚、斯洛伐克和塞尔维亚等部分。匈牙利人是公元第一个千年末期侵犯欧洲的部落民族,由七个部落组成,其主要部落马扎尔(Megyeri)的统治者创建了阿尔帕德(Árpád)王朝。阿尔帕德大公伊斯特万(István),在 1000 年受洗为基督徒,并获加冕为匈牙利国王。他监督匈牙利皈依基督教,后来被追认为圣人,即匈牙利的守护神圣斯蒂芬。⑩
匈牙利的王朝斗争消耗了君主政体,使之变得愈益孱弱,结果就是持续的寡头统治。随着部落财产共有制的瓦解,匈牙利君主政体最初拥有甚多地产,再加上来自皇家矿产的收入,其资源可与法英国王媲美。到贝拉三世(Béla Ⅲ,1148—1196)统治的晚期,国王开始分赠皇家地产、属下各县的大片土地、关税、市场收入等。这些不是西欧那样换取服务的封建赠与,而是新兴男爵阶层手中的自由财产。贝拉三世的继承者们为权力斗争继续向贵族竞相分送皇家礼物。⑪
这就是 1222 年国王安德鲁二世签署金玺诏书的背景(参看第 22 章)。⑫它实际上是限制国王权力的宪法文件,只是来自颇为不同的社会参与者。在大宪章的案例中,强大的英国男爵代表整个王国发言,迫使国王约翰限制自己享有的权力。迫使国王签署金玺诏书的不是匈牙利男爵,而是皇家和城堡要塞的军人。实际上,他们想要国王保护自己免受男爵的掌控。⑬匈牙利教会获得格里高利之后强大罗马教皇的支持,也是要求政策变化的重要政治参与者。教会想保护自己的土地和特权不受进一步的侵蚀,也要求把穆斯林和犹太人的商人驱逐出国,让基督徒商人取而代之。金玺诏书的政治运作显示,匈牙利社会已在国家之外组织成强大的竞争团体,包括男爵或上层贵族、士绅、神职人员。⑭
中央权力软弱的第一后果是蒙古人对匈牙利的掠夺。后者征服俄罗斯后,在 1241 年入侵匈牙利。⑮国王贝拉四世试图加强自己的力量,所以邀请大批异教库曼人(Cuman)进入匈牙利,反而激怒自己的贵族,后者因此拒绝参战。库曼人最后也没参战,匈牙利部队在蒂萨河之战(Battle of Mohi)中遭到彻底摧毁。蒙古人占领整个国家,得知大汗在蒙古过世消息之后,方才撤退。
匈牙利在军事上的薄弱促进了国家建设。⑯匈牙利不知道蒙古人何时归来,也不知道还有没有其他的东方入侵者。为未来威胁作准备,路易一世(Louis I)等的后续国王投入实质性的军事行动,以扩充对巴尔干半岛的控制,甚至抵达遥远的那不勒斯。国家还实施很多改革,以保护自己免受侵略。这包括建造大量石堡和城防,以替换顶不住蒙古进攻的木砖建筑;还以西欧模式的重甲骑士,取代匈牙利军队的轻骑兵。
军事压力导致匈牙利国王促进士绅的利益。然而,这类军人和官员没有直接进入中央国家的架构。后来的软弱国王允许他们为强大男爵服务,促使单一贵族阶层涌现。到 14 世纪,当初促成金玺诏书的皇家和城堡要塞的军人发现,自身的利益不在国王一边,而在男爵一边。⑰
结果是非常软弱的国家,以及寡头地主团体所控制的强大社会。包括新近获得爵位的士绅的匈牙利贵族阶层,完全拥有自己财产,不欠国王任何服务义务。阿尔帕德朝代在 1301 年结束之前,国王虽是当选的,却只是个装饰。他手下没有重要的军队或资源,也没有强大的中央官僚机构。后继的安茹(Angevin)王朝治下,分权过程得到暂时逆转。该王朝终结于 1386 年,贵族迅速卷土重来。
一直到 16 世纪末,莫斯科公国的创始家族,持续成功地孕育男性继承人。这大大帮助了其强大国家的兴起,再一次显示人类制度的偶然性。相比之下,匈牙利却面对重复的继承权斗争,因为它的朝代短命,很多国王又有外国出身。⑱觊觎王位者为了争得权力,只好让贵族得到资源好处。在西吉斯蒙德(Sigismund)国王治下,很多国王城堡都落到了贵族手中。⑲
事实上,匈牙利贵族以议会形式将自己的权力制度化,其权力超过法国的高等法院、西班牙议会、俄罗斯的缙绅会议。⑳远远早于约翰·洛克,贵族阶层“宣布他们有权保护王国的福祉,甚至可以反对国王,如果他试图损害共同利益”。他们还以此理由监禁一名国王。㉑召开议会的先例可追溯到金玺诏书的时代,到 15 世纪中,国家议会每年开会,有权选择国王。不同于英国议会,匈牙利议会受大地主贵族的控制,仅仅代表贵族阶层的利益。如历史学家派尔·恩格尔(Pal Engel)所说,“新制度的本质是决策权的分享,在理论上分给王国中所有地主,在实践中只给参与政治的贵族”。㉒早些时候,城市也可以参加议会,但随着其影响的式微,而逐渐中止。㉓(图 3 显示中世纪匈牙利的政治权力分配。)
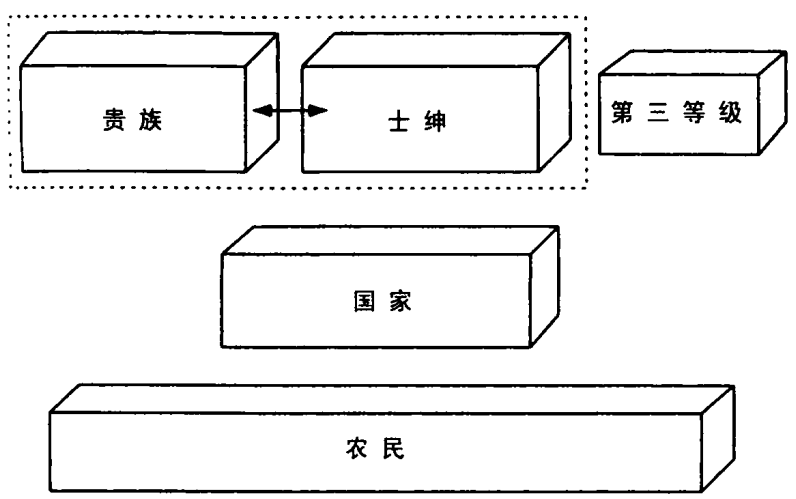
匈牙利打造强大国家的最后机会是在 15 世纪下半叶,奥斯曼帝国已在东南方蠢蠢欲动。贵族地主约纳斯·匈雅提(Janos Hunyadi)在 1446 年被议会推选为摄政王。他通过一系列对土耳其人的军事胜利,包括 1456 年英勇的贝尔格莱德保卫战,而获得巨大威望。㉔约纳斯儿子马蒂亚斯(Mátyás,或写作 Matthias Corvinus)在 1458 年因此而当选国王,在长达三十多年的统治期间,完成了中央国家的现代化。这包括创建国王直接控制的强悍的黑军(Black Army),以取代纪律不佳和半私人的贵族部队,后者曾是匈牙利军队的基础;发展皇家秘书处,并配以大学培养的官员,以取代家族化的贵族官员;实施全国海关和直接税赋,中央政府征税急剧上升。㉕马蒂亚斯使用这些权力新工具,在波斯尼亚和特兰西瓦尼亚(Transylvania),取得了对土耳其、奥地利、波兰和西里西亚的重大军事胜利。㉖
因军事上的必需,马蒂亚斯投入其他现代化专制君主也在做的努力。不同于法国和西班牙的国王,他仍需面对强大和组织良好的贵族阶层,被迫定期向选他当国王的议会征求咨询。贵族因他的军事成功而给他活动余地,但怨恨他所强加的与日俱增的税赋,以及在决策中自身影响力的销蚀。马蒂亚斯死于 1490 年,贵族收回中央国家在前半世纪争得的大部分权利。他们愤愤不平于自己特权的损失,渴望恢复旧状。所以,男爵们将一名软弱的外国君主推上王位,删减黑军的经费,然后将之送上土耳其的战场,结果遭到歼灭。贵族阶层还以国防能力为代价,将自己的税收负担降低 70% 到 80%。
匈牙利返回到贵族分权的均势,很快承受后果。纪律散漫的贵族部队在 1526 年的莫哈奇战役中被苏莱曼一世打败,匈牙利国王被杀。相互争执的男爵只顾反对国家,不顾国家防御;如此场景曾在蒙古入侵中发生,现又重演。匈牙利失去独立地位,一分为三,分别受奥地利哈布斯堡、奥斯曼、土耳其属国特兰西瓦尼亚的控制。
自由和寡头政治
我详细讨论匈牙利的例子,是为了挑明一条相对简洁的见解:强大、凝聚且武装齐全的民间社会,能抵制中央政府,但不一定能获得政治自由。即使有宪政安排,对行政权力实施严格的法律限制,也不能保障政治自由。匈牙利正好符合上述的一切,它得以削弱中央权力,以致国家都不能抵御迫在眉睫的外国敌人。波兰身处类似情境,软弱的国王受贵族会议的控制。两个世纪之后,波兰步匈牙利的后尘,也失去国家的独立地位。
国家独立的丧失不是匈牙利失去的唯一自由。毕竟,匈牙利面对的是庞大和组织良好的土耳其帝国,后者已在欧洲的东南部兼并了多数王国和公国。即使是一个更为集权的现代国家,恐怕也承受不了土耳其的冲击。匈牙利中央国家的脆弱,使匈牙利农民和城市处于从属地位。蒙古入侵带来了动乱和人口骤降。这之后,农民基本上成为自由人,尤其是住在皇家领地上的。作为皇家“客人”,他们有固定的权利和义务,既可充任士兵,又可以缴税来替代。他们最重要的自由是流动自由,并可选举自己的法官和教士。㉗
但世俗和教会的地主都想把他们绑在土地上,成为可供交易的商品。皇家土地分给私人始于 13 世纪,结果使更多农民陷入地主的司法权和掌控。始于 16 世纪早期的食物涨价,促使地主增加农民所欠的领地实物税。农民被迫从事更多的劳役,从前一个世纪的每星期一天,到 1520 年的每星期三天。农民选择地方法官和教士的权利也很有限,需要接受领主的控制。㉘此外,地主开始阻止农民在不同领地之间自由搬迁,或阻止他们移往城镇。日益恶化的境况导致了 1514 年的农民大起义,起义遭到残酷镇压。起义领袖被架在火堆上“登基”,他的同伴被迫分吃从他身上割下来的肉。㉙这次起义发生在土耳其人入侵的前夕,为奥斯曼帝国的胜利贡献了有利条件。㉚
如本章开头所提到的,愈益剧烈的农奴制回潮并不局限于匈牙利。它也发生于波希米亚、波兰、普鲁士、奥地利和俄罗斯。整个地区的贵族要求增强税赋,取消自由,限制属下人口的流动。20 世纪教诲我们,把暴政视作强大中央国家的行径,但它也可来自地方上的寡头。在当代中国,侵犯农民权利、违反环保和安全法律、从事肆无忌惮的贪污,其中最恶劣的案例,多是地方党员干部所为,或是受他们庇护的私人雇主所为,与北京的中央政府无关。中央政府的责任,就是以执法来抑制寡头;有时失去自由,不是因为国家太强大,而是太软弱。第二次世界大战后的美国,吉姆·克劳法(Jim Crow law,编按:泛指 1876 年至 1965 年间美国南部各州及边境各州对有色人种[主要针对非裔美国人,但同时也包含其他族群]实行种族隔离制度的法律)和种族隔离持续二十年,其终止还要靠联邦政府在南方各州强制执行宪法。由此看来,赢得政治自由,不是国家权力受到遏制时,而是强大国家遇上同样强大社会的制衡时。
美国创始人理解此种平衡的必需。亚历山大·汉密尔顿(Alexander Hamilton)在《联邦论》第 17 篇中,描述州政府和联邦政府之间的权利分享。他说:
君主最终战胜属臣的案例中,其成功主要原因是属臣对其手下实施暴政。男爵,即贵族,既是君主的敌人,又是平民的压迫者。君主和平民对男爵又怕又恨,直到相互的危险和利益促成他们联合起来,那就会置贵族权力于死地。如果贵族以仁慈和公正保住其侍从和追随者的忠心耿耿,在与君主的竞争中,几乎永远是赢家,从而削弱或颠覆君主的权力。
汉密尔顿继续说,联邦架构中的州政府就像封建社会的男爵,它们独立于中央政府,其独立程度则取决于如何对待自己的公民。强大的中央政府在本质上是不分好坏的,其对自由的最终影响,取决于它与从属权力机构的互动。这是美国历史上的真理,也在匈牙利和波兰的历史上体现出来。
另一方面,如果强大国家与强大寡头相互勾结,自由就面临尤其严峻的威胁。这就是俄罗斯所处的情境,莫斯科公国在匈牙利国家消亡的同一世纪崛起。
第 26 章 更完美的专制主义
莫斯科国家的涌现和俄罗斯政治发展的特征;君主政体依赖贵族,造成俄罗斯农民逐渐沦为农奴;与欧洲其他地区相比,专制主义在俄罗斯取得更为彻底的胜利
尤其在弗拉基米尔·普京兴起的 21 世纪初,俄罗斯联邦成为政治学家所谓的“选举式威权”政权。①政府基本上是威权的,受控于政治家、官员和商业利益所组成的灰色网络,但仍然举行民主选举,使继续执政获得合法性。俄罗斯民主的质量很低。政权控制几乎所有的主要媒体,不允许对政府的批评,威胁反对派候选人,或使之丧失参选资格,还向自己的候选人和拥护者提供优惠。
它在法治上的表现,比民主质量问题更为糟糕。揭露官方腐败或批评政府的新闻记者突然死去,没有看到找出杀手的真正努力。私人企业遇上政权内线人士的敌对接管,便会遭到政府部门的诬陷指控,从而被迫放弃资产。不夸张地说,重要官员即使参与谋杀,也可逍遥法外,无须负责。专门调查世界上腐败水平的非政府机构“透明国际”(Transparency International),将俄罗斯排在 180 个国家中的第 147 名,劣于孟加拉、利比里亚、哈萨克斯坦、菲律宾,稍稍优于叙利亚、中非共和国。②
很多人看到 21 世纪的俄罗斯和前苏联的连贯性,又因俄罗斯人经常表达对斯大林和苏联岁月的怀旧,而得到放大。布尔什维克革命后的七十年,共产主义扎根于俄罗斯,当然塑造了当代俄罗斯人的态度。
但在共产主义的下面藏有很多龟。如果仅把当代威权主义归罪于 20 世纪的政治,首先就要解说,共产主义为何在俄罗斯和中国获得彻底的成功。当然,发挥作用的还有更古老的专制传统。布尔什维克革命之前,俄罗斯已发展了强大的中央国家,其行政权力只受法治或负责制立法的软弱约束。布尔什维克之前的俄罗斯,其取得的专制主义的性质,不同于法国或西班牙的旧政权,更接近于前现代中国或奥斯曼。个中的原因与俄罗斯的地理环境有关,地理环境对它的政治文化产生了持久影响。
俄罗斯专制主义的来源
公元第一个千年末期,俄罗斯国家起源于乌克兰的基辅地区,基辅曾是主要的贸易站,连接北欧和拜占庭帝国等。它的持续存在中断于 13 世纪 30 年代末,其时,拔都可汗和速不台率领的蒙古军队侵占俄罗斯,基辅遭到彻底摧毁。身为教皇使节的大主教迦儿宾(Carpini)写道,经过基辅时,“我们看到现场有无数死人的头颅和骸骨,该城曾经很大,人口众多,现在却一片荒芜,只有两百栋房子还立在那里,俘虏在从事着最为恶劣的苦役”。③蒙古占领持续了将近二百五十年。很多当代俄罗斯人,被问到为何他们的国家和政治文化迥然不同于西欧时,立即把责任推到蒙古人身上。西方也有观察俄罗斯的悠久历史,如侯爵德·屈斯蒂那(de Custine)。他坚持俄罗斯应被视作“亚洲”强国,它与蒙古人、奥斯曼人、库曼人和其他亚洲人的互动,对它的成型发挥了决定性作用。④由于蒙古人民共和国的出现,见解已经转变。新一轮修正主义的历史评论,以更为肯定的语气解说蒙古人的作用。⑤
不管怎样,蒙古入侵对俄罗斯后续的政治发展施加了重大影响,且多半是消极的。⑥首先,它切断了俄罗斯与拜占庭和中东的贸易和思想交流,后者曾是俄罗斯宗教和文化的来源。也阻碍了它与欧洲的联系,这意味俄罗斯没像西欧那样,深入参与相关的历史进程,比如文艺复兴和宗教改革。
其次,蒙古占领大大延误了俄罗斯的政治发展。基辅遭到彻底摧毁后,基本上需要重头开始,当代乌克兰的基辅地区曾是俄罗斯老祖宗的定居点。蒙古人抵达之前,俄罗斯国家已经开始分裂。政治权力向无数自称为王的小封地流散,又因蒙古占领而获得确认。俄罗斯的重心从黑海北部的欧洲本都地区(pontic)转向东北部,当地的莫斯科大公国崛起,成为政治舞台上的中心角色。不像持续八百年的欧洲封建主义,俄罗斯的封地仅生存两个多世纪——从 1240 年开始套上鞑靼轭(Tatar yoke)到伊凡三世当政的 16 世纪中期——很快,小封地的领主必须面对日益强盛的中央君主政体。
最后,蒙古人破坏了继承于拜占庭的法律传统,使政治生活变得更为恶劣、更为残忍。与欧洲的基督徒君主相比,蒙古统治者把自己看作纯粹的掠夺者,其目的就是从所控制的居民身上榨取资源。他们仍处在部落层次,从未发展出政治制度或正义理论,可以带给所征服的居民。他们不像传统农业国家的统治者,并不矫饰自己是为被统治者而存在的。他们只有很短的时间表,愿意以不可持续的方式大规模榨取资源。他们严惩任何抵抗力量,为了杀鸡儆猴,甚至愿意处死整座城镇的居民。他们招募俄罗斯的领主成为自己的征税官,包括将来创建俄罗斯国家的莫斯科大公。他们以自己的掠夺策略,训练数代的俄罗斯领袖。事实上,他们通过联姻而融入俄罗斯人口的基因。
像我们所讨论的几乎所有政治体,发动战争的需要促动了俄罗斯的国家建设。像基于法兰西岛的卡佩家族,莫斯科的留里克王朝(Rurik)从自己的中心位置向外扩展,征服和吸收其他封地公国、蒙古和立陶宛等的外国军队。伊凡三世(1440—1505)治下的国家成为重要力量,兼并诺夫哥罗德和特维尔(Tver)。他给自己冠上全俄罗斯大公的称号,莫斯科公国从伊凡一世(1288—1340)的六百平方英里,到瓦西里二世(1415—1462)的一万两千平方英里,再到他自己统治结束时的五万五千平方英里。⑦
封地期间的俄罗斯国家,其形成非常类似于中国和奥斯曼的国家形成过程。像中国西周的创始朝代,基辅贵族家庭的后裔分布于俄罗斯各地,尤其是在蒙古入侵之后。他们组建一系列小公国,相当于俄罗斯版本的封建主义。每位领主控制自己的领地、经济资源和军队,并与自由贵族(boyar)签订契约以获服务。
莫斯科国家的权力基于服役贵族(middle service class),由骑兵组成,报酬不是现金而是封地(pomest’ia),每块封地上约有五或六户农家。由于地多人少,控制人口比控制土地更为重要。骑兵不是常备军,受领主召集而提供服务,军事季节结束后,再回到自己封地。俄罗斯和奥斯曼的封地非常相像,这可能不是意外。其时,俄罗斯与土耳其的接触愈益增多。像奥斯曼的骑士,俄罗斯部队的核心成员,如果身处欧洲其他地区,便被称作低层士绅,其土地和资源全部来自国家。俄罗斯骑兵配置相对轻便的装备,主要倚靠迂回战术。这很像奥斯曼骑兵,而迥然不同于西欧的重甲骑士。莫斯科政权组建此种部队的动机,也与奥斯曼相似。这个军事组织的地位全靠国家,不会要求现金军饷。它可被用来抵消领主和贵族的势力,后者拥有自己的土地和资源。⑧
这是俄罗斯和匈牙利的重大差异。在俄罗斯,服役贵族接受招募,直接为莫斯科国家服务。在匈牙利,它变成贵族阶层的一部分。这不同选择也许足以决定后来的分道扬镳,一个社会走上中央集权,另一个趋于权力下放。与西欧社会相比,俄罗斯社会对莫斯科的国家建设设置了较少障碍,原因之一就是:服役贵族直接从属于国家,没有接受领土贵族的再次分封。
俄罗斯版本的封建主义历史太短,尚没达到根深蒂固的程度,这是俄罗斯贵族无法限制中央国家权力的另一原因。俄罗斯是否经历过封建主义?俄国史学界对此有长期争论,因为俄罗斯的封地从没获得西欧对应物所享受的自治权。⑨俄罗斯的领主和较低层次的贵族没有时间建造城堡,俄罗斯的平原和大草原,将优势赋予快速移动的进攻军队,而不是防御军队。
莫斯科国家颁布门第选官制(mestnichestvo,编按[下同]:俄语为Местничество),故意在贵族中播种不和。它将贵族家庭以及家庭内的个人划出等级。像法国和西班牙的爵位和特权的出售,门第选官制也让贵族互相竞争,从而破坏了贵族内部的凝聚力。⑩所以,俄罗斯贵族作为一个阶层,其内部团结很差,几乎没有发展出联合抵抗中央国家的机构。他们以内部的小争执而著称,经常自我损耗。
在俄罗斯,法治一开始就比西欧薄弱。天主教会在领土主权国家之外建立教会法规,但俄罗斯东正教从没扮演过类似角色。被俄罗斯当作模型的拜占庭帝国,其教会和国家的关系是政教合一。东罗马皇帝委任君士坦丁堡的牧首(Patriarch,最高主教),裁决教会中的教条争议。格里高利改革和叙任权斗争的相似情形,从没在拜占庭的世界发生。东正教没有发展出可颁布法律的国家般的中央机构,也没像天主教会一样,将牧首法令编纂成统一的教会法规。当蒙古入侵切断了俄罗斯教会与拜占庭的交往时,它在莫斯科国家身上找到新监护人。教会和国家的利益相互吻合,后者提供赞助和权力,前者鼓吹后者作为“第三罗马”的合法性。大牧首尼康(Nikon)在 1666 年遭到开除,之后的俄罗斯教会彻底变成政教合一。到 1721 年,彼得大帝颁布《宗教事务管理章程》,干脆取消牧首职位,取而代之的是沙皇指定的神圣宗教会议(Holy Synod)。⑪
如果怀疑法治保护西欧精英的重要性,我们只要想想所谓的沙皇特辖制(oprichnina,俄语опричнина)。那是俄罗斯历史上的黑暗年代,时值伊凡四世(1530—1584)统治的后半期,在西欧历史中找不到对应物。他被后人称作伊凡雷帝(Ivan Grozny,俄语Иван Грозный),既可译作恐怖伊凡,也可译作伊凡大帝。他心爱的年轻妻子阿纳斯塔西娅(Anastasia,俄语Анастасия)死于 1560 年,使他对周遭的宫廷官员疑鬼疑神。他突然离开莫斯科,至 1565 年方才返回,要求贵族让他建立所谓的非常行政区,并让他享有处理恶人和叛徒的唯一大权。一旦获得同意,他就发起恐怖统治,反过来攻击贵族。愈来愈多的贵族与家人一起遭到逮捕、折磨、处决。伊凡创建了所谓的特辖军(oprichniki,俄语опричники),身穿黑衣,骑黑马,成为他法外统治的特殊工具。特辖区内的私人财产遭到国家没收。之后,它又得到扩张,最后面积相当于全国的一半。估计有四千至一万的贵族被杀,古老领主家庭中存活的仅得九家,大部分土地都被充公。⑫伊凡四世好像完全失去情绪平衡,一度致命地伤害了自己的儿子兼继承人。他死后,俄罗斯只能说仍然心有余悸。⑬很难说,它不是斯大林在 20 世纪 30 年代中后期实施党内清洗的先例。其时,苏共总书记怀疑身边处处有阴谋诡计,杀光了当年与其携手闹革命的老共产党员。⑭它也使人不得不忆起清洗贵族精英的中国统治者,像武则天。
从俄罗斯政治发展来看,使人迷惑的是贵族为何授予伊凡这些特权,祸及自身。有人认为,他们不敢想象自己可以独自当政,也害怕君主不掌大权的后果。在伊凡四世奇怪消失于莫斯科的时候,有人提出如此可能。俄罗斯人对软弱国家会造成的混乱和崩溃心怀恐惧,这并不荒谬。他儿子费奥多(Feodor,俄语Фёдор)去世于 1598 年,没有留下子女,留里克王朝因此而告终,开始了所谓的混乱时期。莫斯科国家饱受饥荒和外国侵略的困扰,因一系列“伪德米特里”(false Dmitri,俄语Лжедмитрий)竞争君位而分崩离析。莫斯科君主创造的国家机器不够强大,承受不了漫长的继承权斗争。即使君主权力崩溃,也不能回归到分权的封建统治。结果只是失序的暴力和外国的霸权,直到罗曼诺夫王朝在 1613 年涌现。
自由选择
俄罗斯专制主义的兴起并不由俄罗斯文化内在逻辑所命中注定。事实上,俄罗斯历史上有西方的共和或代议制度的先例,为其他可能性提供视野。西北部的城市诺夫哥罗德从没被蒙古人征服,在早期封地时期,一直是颇具活力的商业共和国。它与波罗的海贸易紧密相连,发挥门户作用,让欧洲货物进入俄罗斯。诺夫哥罗德的君主统领军队,但在执政时受市民大会(veche,俄语вече)的限制。市民大会从城市贵族中选出市长,所有自由公民都可投票。它还控制税赋、法律和外交,甚至可以解雇君主。城市内,社区在料理自己事务上行使很大自治权。诺夫哥罗德最终被伊凡三世征服,在 1478 年成为莫斯科国家的一部分。伊凡三世废除诺夫哥罗德所有的共和机构,将很多当地领袖当作叛徒处死,并将大量贵族和商人家庭驱逐出境。⑮
第二个代议机构是缙绅会议,由贵族组成,近似于法国三级会议和西班牙议会。它的开会并无定律,但在适当时刻扮演重要角色。它批准了伊凡四世的数项倡议,例如他向利沃尼亚的开战。其他会期批准了伊凡四世儿子费奥多在 1584 年的继位,并在 1598 年向摄政王鲍里斯·戈杜诺夫(Boris Godunov,俄语Борис Годунов)提供皇位。它最重要的举动是在 1613 年核准米哈伊尔·罗曼诺夫(Mikhail Romanov,俄语Михаи́л Фёдорович Рома́нов)成为沙皇,从而终止混乱时期。该议会在 17 世纪还继续开会,批准了多次宣战和税赋,直到彼得大帝使之边缘化。⑯自那以后,代议机构在俄罗斯销声匿迹,直到 1906 年日俄战争之后召开的立法机构杜马(Duma,俄语Дума)。
抵制权力的最后潜在来源是俄罗斯教会。如上所述,评论家经常谴责俄罗斯教会是莫斯科统治者的驯服工具,不管是沙皇时期,还是今天。但在大牧首尼康被开除之前的时期,仍有可能走上不同途径。俄罗斯东正教曾拥有近乎四分之一的俄罗斯土地,由此而享受自治。自圣谢尔盖(St. Sergius)改革以来,俄罗斯就有优良的修道院传统,但经常引起世俗统治者的怀疑。至少在佛罗伦萨大公会议(Florentine Union)触发危机之前,莫斯科的都主教(Metropolitan)都由君士坦丁堡的最高主教指派,俄罗斯君主无从置喙,之后才由俄罗斯主教会议选出。⑰也有个别教会领袖不畏暴政,如莫斯科都主教菲利普(Philip),因为谴责伊凡四世,而被赶出自己的教区,最终竟被勒死。⑱
这些案例表明,俄罗斯传统并不是暴政不断,自由选择时有发芽开花。共产主义倒台后重现创造更为自由社会的诺言,但其兑现恐怕还在将来。
农奴所有者结成卡特尔(Cartel)
17 世纪末的俄罗斯国家已有中央集权,但还比不上欧洲对手。没有整齐的中央官僚机构,只有一系列所谓的衙门(prikazy,俄语Приказы),其职责既有重叠,又不一致,来自沙皇繁杂的指令(prikaz,俄语Приказ)。⑲不同于法国的总督制度,从地方到中央的政府任命,都出自沙皇,被称为“给食”(kormienie,俄语Кормление),这名字就表明制度背后的监督和掠夺意味。早在 16 世纪既已存在的地方自治政府,在伊凡四世的治下遭到废除,国家倚靠军事总督制度(voevody,俄语Воевода)来实施行政命令。军队也同样原始,仍然基于骑兵,只在首都组织新型步兵,但不一定靠得住。⑳
俄罗斯国家建设的下一轮是在彼得大帝(1672—1725)治下。他迁都圣彼得堡,又从欧洲引进一大批新制度。彼得是个巨人,不论是体形,还是领导才能,单枪匹马尽力推行自上而下的社会改造。战争再次成为国家建设的主要动力,尤其是对抗瑞典的北方战争(Great Northern War)。彼得在 1700 年纳尔瓦战役中,败于瑞典皇帝查理十二世,遂开始对当时欧洲边界的俄军进行彻底重整,并从零开始打造海军(从最初的单船只舰发展到最终能够战胜瑞典海军的八百艘)。他也推行俄罗斯中央政府的现代化,废除老式衙门,换成模拟瑞典的参政院(a system of colleges)。参政院以技术专长为基础——大多来自外国——在辩论和执行政策方面发挥了特殊功能。
15 世纪和 16 世纪国家建设的第一期,主要是动员服役贵族,这分裂了贵族阶层,确保他们大部分直接依赖国家。彼得甚至更进一步,征召整个贵族阶层参与国家服务。贵族入伍先当小厮,其提升全凭现代的择优标准,一生必须附属于自己的团队。所以,与欧洲相比,俄罗斯贵族服务的观念更为持久,虽然实施方式大相径庭。为国家服务的贵族随身不带自己的属臣和侍从,却在中央等级机构中获得职位。这导致俄罗斯社会的总体军事化,在道德上重视责任、荣誉、等级、服从。㉑
支撑俄罗斯专制主义的内部政治力量,其平衡可用图 4 来说明:
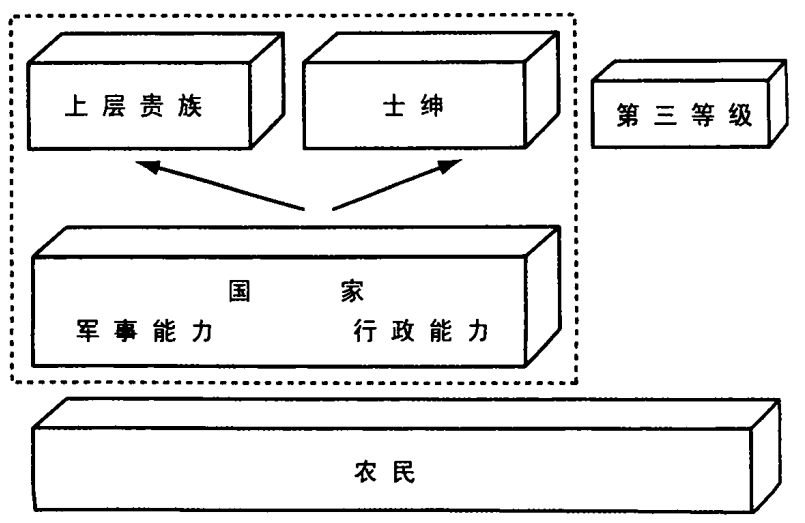
彼得大帝在 1722 年以官秩表(Table of Ranks)替换古老的门第选官制。每个国民都有自己的法定等级,以及相应的特权和义务。非贵族人员一旦升到足够的等级,不管是在官僚机构还是在军队,就可自动进入世袭贵族的行列。新鲜血液进入贵族,这很有必要,因为国家需要大批公职人员。官秩表确定贵族的集团身份,并加强其采取集体行动的能力。但它从不将自己视作君主政体的对手,因为其利益与国家紧密相连。㉒
贵族提供服务,以换取免税、土地人口专有权和进一步榨取农奴的机会。农奴在君主赠与贵族的封地上首次出现,这显示农民条件的恶化和贵族服务阶层的兴起,以及两者的密切相连。这些封地倾向于在南方、东南和西方,都是国家夺之于邻国的新土地。辽阔的北方领土上没有战事,它的农民处境要好得多——基本上只是国家的农民,只有欠国家的义务,不欠私人地主的。㉓
整个 16 世纪和 17 世纪,农民的税赋日益上涨,而更为重要的法律限制则针对农民的行动自由。根据古老的传统,农民有权离去,但在后来受到愈来愈多的限制,最后竟被全部废除。㉔限制农民的迁徙是至关重要的,它直接影响俄罗斯贵族的团结,以及贵族与君主政体的同盟。
讽刺的是,此中原因与俄罗斯的地理有关。它缺乏地理界限,非常不适于奴隶制的发展。俄罗斯只有很少阻挡迁徙的自然屏障,如无法通行的大河或山脉。国家因扩张而不断拉长边境线,尤其是在南方和东南方。乌克兰南部和顿河盆地(Don Basin)的自由哥萨克社区,据说由逃走的农奴所建。像蓄奴农地与开放边境相邻的美国南方,只有农奴主之间达成牢固协议,以限制农奴行动、送回逃奴、既严罚逃奴又严罚违规地主,农奴制度方能取得成功。如有主要参与者不予合作——或是部分地主,或是自由城市,或是向逃奴提供保护的国王——整个制度就会崩溃。考虑到这段时期劳力缺乏,任何地主如果退出联盟,以较好条件将农奴吸引到自己领土,便会获取丰厚的利润。所以,必须以显著的地位特权和严守反迁徙规则的承诺,来加强地主卡特尔的团结。俄罗斯专制主义的基础,就是君主和上下层贵族的同盟。他们都答应遵循有关规则,牺牲品就是农民。
维持农奴卡特尔的需要,解释了俄罗斯政治发展的众多现象。自己没有农奴的个人,欲自由拥有土地,会受到政府愈来愈多的限制。要想得到土地,必须先变成贵族。一旦成为贵族,便能自动获得农奴,以及维持这一制度的义务。此举遏制了资产阶级在独立商业城市的成长。西方的城市在促进农民自由方面扮演了重要角色。因此,在俄罗斯率先发展资本主义经济的是贵族,而不是独立的资产阶级。㉕维持卡特尔的需要,也解释了俄罗斯向南方和东南方的扩张。边境旁边的自由哥萨克社区的存在,无疑是持续的诱惑,也是逃跑农民的良机,必须遭到镇压。
彼得大帝之后
彼得大帝是推行现代化的伟大人物,在很多方面促使俄罗斯“欧洲化”,并使之成为欧洲政坛中的主要角色。但他从上到下的强行改革,遇上了俄罗斯社会本质上的局限。例如,他试图创立省和地区的两级制度,以及新式的市政规则,以改造省、市和地方的政府,到最后都不了了之。用当代发展中国家的字眼来说,原因在于“能力缺乏”。那是指,地方上受过训练的行政人员不够,现存的又缺乏热情。中央颁布的法令得不到实施,政权也无法铲除既有的腐败和独断专行。㉖
彼得大帝在军队和中央官僚机构中,推行选贤与能的现代晋升制度。他死后,便难以为继。他的很多革新全靠自己的监督和精力,例如,他曾旁听政府招聘干部的考试。他去世后,宫廷内外的强大家族使行政机构重趋家族化。他的继位者软弱。想要晋升到文武官职的最高等级,全得倚靠豪门巨室的赞助,像多尔戈鲁科夫(Dolgorukov,俄语Долгоруков)、拉雷斯金(Naryshkin,俄语Нарышкин)、戈利岑(Golitsyn,俄语Голицын)、萨尔蒂科夫(Saltykov,俄语Салтыков)。日益掌控国家政策的贵族在 1762 年废除自己的服务义务,并获得更多针对农民的权利,如随意迁徙和驱逐农民的权利。㉗豪门家庭及其赞助网络的相互竞争,甚至延伸到军队,战斗力因此而受损。
贵族家庭的兴起分散了俄罗斯制度的权力,并软化伊凡四世和彼得大帝所遗留下的专制传统,再加上法国文化在俄罗斯精英中所享有的主导地位,这一切使托尔斯泰《战争与和平》所描述的 19 世纪早期的贵族社会,看来酷像欧洲的贵族社会。如在两百年之前,这是不可想象的。但这种权力分散与西方现代行政国家的兴起,不可混为一谈。历史学家约翰·勒多内(John LeDonne)说:“全国范围家族和依附者的网络,无不在嘲弄立法文件所建立的严格等级制度。此类立法努力,一直在寻找行政秩序和‘规范化’。它解释了为何俄罗斯政府,比任何其他政府都更是人的政府,而不是法的政府。”㉘
专制主义完成
有关俄罗斯的解说,以 18 世纪晚期稳固专制国家的出现而告结束。显而易见,之后又有很多新的发展,包括 19 世纪的自由派实验和 20 世纪极权国家的兴起。到法国大革命时,俄罗斯统治的特征已昭然若揭,它既不同于法国和西班牙的弱的专制主义,也不同于中国和奥斯曼的国家。
在若干方面,俄罗斯的国家比法国或西班牙更为强大。至少在与精英打交道时,后者受到约束,必须尊重法治,这在俄罗斯却是闻所未闻的。法国和西班牙的政府,以债务违约和货币操纵来蚕食产权,甚至捏造指控,通过法律程序来勒索钱财。但至少,他们觉得必须运用现有的法律机构。相比之下,俄罗斯政府无需法律借口来没收私人财产,逼迫贵族为政府服务,处置敌人和叛徒时,漠视正当的法律程序。伊凡四世的特辖制在某种意义上只是意外,之后也没有相似的复制,直到 20 世纪的共产主义政府。但它的曾经发生,为后来俄罗斯统治者创立了重要的先例。他们很清楚,他们手中针对精英的极端措施是西方主权国家所没有的。在这一方面,俄罗斯政府更接近帝制中国,更远离西方。俄罗斯政府发展了类似奥斯曼的专制制度,譬如作为骑兵报酬的封地。奥斯曼和马穆鲁克,即使在最兴盛时期,也比俄罗斯统治者更加尊重法治。
另一方面,俄罗斯的专制主义更为家族化,远远超过中国或奥斯曼的版本。如我们所见,中国人发明了现代官僚机构和非人格化的中央统治。大体上,中国历史是国家与家族制复辟的斗争史。早在中国统一之前的公元前 3 世纪,非人格化和选贤与能的政府的理想就已问世。奥斯曼的军事奴役制建立任人唯才的行政制度,不受家族影响,在其全盛时期,不乏来访欧洲人的赞美。彼得大帝想在俄罗斯创建同样制度,只取得部分的成功。俄罗斯的家族力量随后轻易夺回政府,以不透明的方式在幕后制定政策。
当代俄罗斯,与彼得大帝死后的百年社会有惊人的相似。尽管有现代的正式宪法和书面法律,俄罗斯国家仍受灰色精英网络的掌控,很像曾经控制帝制俄罗斯的萨尔蒂科夫和拉雷斯金家族。这些精英行使权力的方式,不是法律或规范程序所能定义的。但与中国不同的是,俄罗斯最高精英没有对国民负责的类似道德感。在中国,政治等级越高,政府质量越有改进。但在俄罗斯,它却变得越糟。当代精英愿意借用民族主义,使自己的权力合法化,但到最后,好像仍在为己着想。 俄罗斯没有陷入历史的泥潭。伊凡四世、彼得大帝、斯大林开下专制先例,但接踵而至的却是自由化。今日社会已被动员起来,其方式不同于旧政权时期,资本主义的引进允许精英的组成定期更换。今日腐败和紊乱的选举式威权主义,不再是俄罗斯人曾承受的残酷独裁。俄罗斯历史提供很多通向较多自由的其他选择,可作为将来改革的先例。
第 27 章 征税和代表权
失败的负责制案例,帮助理解议会制度在英国的发展;政治团结的来源,其在诺曼征服之前英国的扎根;英国制度合法化中的法律作用;光荣革命所真正实现的
政治负责制如何发展的最后案例是英国,其政治发展的三大组件——国家、法治和政治负责制——都成功获得了制度化。我最后审视英国是为了避开辉格史观的缺陷。关于英国代议政府的兴起,已有很多论述认为,它是可溯源自古代雅典的西方发展模式的逻辑的、必然的或无可避免的结果。但这些论述很少互作比较,所引证的一系列因果事件,又忽略了不易察觉或更为遥远的因素。而在事实上,那些因素却在扮演重要角色。换言之,它们只看到顶部的龟,而忽视了蛰伏于下的龟。
我们得以避免这个问题,因为我们已讨论四个负责制政府无法出现的欧洲国家——如果把所讨论过的非西方案例也包括在内,那就不止四个。我们观察英国与其他案例的异同,将更好地了解促使负责制发展的组合因素。
跟法国、西班牙、匈牙利和俄罗斯一样,英国首先是部落社会,然后是封建社会,它的中央集权始于 16 世纪晚期和 17 世纪早期。这些社会的精英都组成政治团体——英国议会、法国高等法院、西班牙议会、匈牙利议会、俄罗斯缙绅会议——推行现代化的君主要向它们寻求支持和合法性。在法国、西班牙和俄罗斯,这些团体没能凝聚成强大的制度化参与者,没能对抗中央国家,没能取得宪政上的妥协,没能获得国王对自己的负责。相比之下,英国议会却是强大而凝聚的。
具体地说,不同于主要代表卡斯提尔城市的西班牙议会,或贵族掌控的法国和俄罗斯的政治团体,英国议会不仅代表贵族和神职人员(世俗和精神的领主),而且代表广泛的士绅、市民和业主。这些平民是议会的灵魂和动力。英国议会强大到成功击败国王的诸多计划,包括增税、组建新军、躲避普通法。它还创建自己的军队,在内战中打败国王,将之处死,迫使继任君主詹姆士二世退位,拥戴来自欧洲大陆奥兰治的威廉(William of Orange)。到最后,统治英国的不是欧洲大陆那样的专制君主,而是正式承认议会负责制原则的立宪君主。英国议会获得如此进展,而欧洲其他地区的议会却四分五裂和软弱无能,或被拉拢收买,或主动支持君主专制,直到法国大革命前夕。有人自然要问,这是为什么?
英国还在另一方面为当代发展中国家树立先例。17 世纪初,早期斯图亚特治下的英国不但日益专制,而且非常腐败。渗透法国和西班牙政府的实践,如卖官鬻爵和家族攫权,同样也发生在英国身上,只是在规模上还算适中。但到该世纪末,英国的公共腐败问题,即使没有得到解决,至少已有很大收敛。政治制度得以废除公职买卖,建立现代官僚机构,提升国家整体的力量和效率。这虽然没有彻底解决英国公共生活中的腐败问题,但阻止国家陷入最终摧毁法兰西王国式的腐朽泥潭。今天,面对普遍公共腐败的发展中国家,可以借鉴英国政治制度的应对方法。
英国政治团结的根源
我们看到,法国、西班牙和俄罗斯的君主政体使用种种策略,在贵族、士绅和资产阶级当中,收买、胁迫、化解潜在的对手。英国君主作出同样的尝试,但议会所代表的社会阶层团结起来,抵制并最终打败国王。问题在于,这团结来自何处。
答案至少有三,有的已在以前章节中获得详细解释。第一,很早以来,英国社会团结的政治性大于它的社会性。第二,普通法和英国法律制度的合法性获得广泛认同,业主保卫自己财产的意愿强烈。最后,此时的宗教,虽然在英国人中间造成痛苦的分裂,却赋予议会超越的使命感。如果与国王的争执只是为了财产和资源,该使命感便不复存在。
地方政府和团结
我们在第 16 章中提到,欧洲的部落社会组织因基督教的影响而趋于崩溃,远远早于现代国家建设。英国在这一过程中,比任何其他地方走得都要快。6 世纪晚期坎特伯雷的圣奥古斯丁传教开始,更加个人主义的社区便取代了扩展的亲戚关系。(这并不适用于爱尔兰人、威尔士人和苏格兰人。他们的部落关系——例如高地氏族——持续到很晚的历史后期。)邻居之间毫无关联,这样的社区在诺曼入侵之前的盎格鲁—萨克逊时期已属司空见惯,使当地农业社会截然不同于东欧,更不同于中国和印度。①
基于亲戚关系的社会组织虽然孱弱,但并不排除社会团结。紧密相连的亲戚团体,可在团体范围内提供集体行动,但遇上宗族或部落之外的合作,又会变成障碍。基于亲戚关系的社会,其集体行动的范围非常狭窄,所以需要政治制度。
英国社会早期的个人主义,并不意味社会团结的消失,只是团结形式是政治性的,而不是社会性的。诺曼征服之前,英国分成相对统一的各郡(shires),它们可能曾是独立的小王国,现已聚集成更大的英格兰王国。主持郡务的是称作长老(ealdorman)的古老官员,其职位是世袭的。(它的词根来自丹麦,现在仍存活于美国的地方政治,市府参事即写作 alderman。)②但实际权力渐渐落到皇家官员手中,即郡治安官(shire reeve,or sheriff),后者受国王的指派,代表皇家权力。每半年,他组织一次郡会议,该区所有自由民(后来变成自由地主)必须出席。③诺曼征服并没摧毁该统治制度,只是将郡改为县,以符合欧洲大陆法兰克人的习惯。然而,治安官的权力大增,取代了世袭的长老。郡会议演变成县法庭,用弗雷德里克·梅特兰的话说:“国王的大领主必须在法律面前人人平等的基础上,与自己的属臣相聚。租户可能与自己领主坐在一起,俨然像个同等伙伴。”④
今天,这些制度的详情好像只有考古价值,但在解释议会作为政治制度的演变时却非常重要。欧洲大陆封建主义的性质,尤其是在卡洛林帝国地区,看来非常不同。欧洲领主贵族享有对司法权的控制,其程度远远超过英国。⑤在英国,国王享有优势。诺曼征服之后,国王利用县法庭来监察封建法庭。如个人觉得在领主那里得不到正义,就可向治安官提出上诉,要求将诉讼移至县法庭。后来,国王法庭(详见第 17 章)取代县法庭成为重要案件的预审庭。后者只得主持较不重要的诉讼,譬如金额不超过四十先令的土地纠纷。与欧洲大陆相比,英国的非精英更有机会运用这些机构。
县法庭开始失去其司法功能的同时,却获得新的政治功能,成为更广泛政治制度的代议场所。正如梅特兰所说:
到 13 世纪中期,我们发现,民选代表被召集来参加全国会议,或叫议会(parliament)。他们是县法庭的代表,不是无组织群体的代表。我们几乎可称他们为集团代表。理论上,整个县都由县法庭代表……国王的巡回法官不时来访,整个县的地主团体(totus comitatus),前来晋见,报告上次来访之后的所作所为。县法庭可作出裁决,也可作证,如有犯错,还会被罚款。⑥
所以,县法庭是奇怪的组织,既自上而下,又自下而上。它由国王所创建,受由国王任命并对国王负责的治安官统辖。但它又以全体地主的广泛参与为基础,不受世袭等级和封建地位的限制。治安官反过来又受地方民选督察官(coroner)的制衡,民选督察官应代表县民利益的观念因此而获得合法性。既有对国王的向上负责,又有对县民的向下负责,两者日益趋于平衡。
郡或县下面还有更小的地方行政单位,叫作百户(hundreds),相当于卡洛林帝国的乡(centenae)。(这些行政单位也传到美国。)百户区有自己的聚会,叫作百户法庭,开始在司法方面扮演日益重要的角色。百户区由治安官任命的巡警所治理,一起负责警察功能,如抓捕罪犯。百户也是英国陪审团制度的基础,需要提供审判刑事案件的十二名陪审员。⑦
因此,甚至在诺曼征服之前,整个英国社会已组建高度参与的各式政治单位,一直抵达村庄层次。这不是地方社会组织参政的基层现象,而是全国政府邀请地方上的参与,构建地方上的生活,扎根成为社区的来源。
普通法和法律机构的作用
值得注意的是,后来英国政治代议制度的构成部件,一开始只是司法机构,像县法庭和百户法庭。英国历史上,法治的出现远早于政治负责制,后者又始终与保护法律密切相联。英国司法的参与性质,加上普通法因应地方需求以定规则的特征,帮助造就了法律属于大家的感情,其强烈程度远远超过其他欧洲社会。公共负责制首先意味着对法律的服从,尽管那时的法律,不论是法官作出的,还是颁成文本的,都没有走过民主政治的程序。
法治的主要功能之一是保护产权。在这一点上,英国普通法比其他地方的法律更为行之有效。正如哈耶克所说的,原因之一在于普通法是分散决策的产物,能尽量适应各地的情形和知识。不过吊诡的是,原因之二在于国王愿意在产权上支持非精英对贵族的反抗,这便需要强大的中央国家。在英国,原告早就可以将产权诉讼移至国王法庭,如金额不够,仍可移至县法庭或百户法庭。中世纪有不少复杂的传统产权,如佃权(copyhold)。土地在技术上是领主财产,但佃户(villein)又可将之传给儿子或亲戚。国王法庭倾向于反对领主,保护佃权所有人的权利,以致这种财产渐渐进化成真正的私人财产。⑧
县和百户层次的法庭众多,国王在地方产权争执中愿意充任中立仲裁人,这一切大大增强了英国产权的合法性。⑨到 15 世纪,英国司法制度的独立性和获得认可的中立性,允许它扮演日益重要的角色。它变成真正的“第三分支”,有资格裁决宪法问题,如议会废除专利特许证的权利。有评论家指出,“很难想象,此类问题能在中世纪欧洲的其他地方获得解决——并且是完全独立的解决——全靠法官以专业语言作出讨论,而不是政治上的樽俎折冲,或有关团体的胁迫。”⑩今天的发展中国家,仍缺乏如此的司法才能和司法独立。
到了 17 世纪的重大宪政危机时,不让君主破坏法治成了保卫英国自由的呐喊和议会团结以抗国王的源泉。出现于早期斯图亚特(1603—1649 年)的威胁是国王的星室法庭(Court of Star Chamber,其起源和司法权都很模糊),其为了“更有效地”起诉犯罪,而省去一般法庭的正常保护程序(包括陪审团的审讯)。在第二任斯图亚特国王查理一世(1600—1649)的治下,它带有更多政治性,不只是起诉犯罪,还用来对付假想的国王之敌。⑪
英国法律独立的更佳象征,莫过于爱德华·柯克爵士(1552—1634)。他是法学家和法律学者,最终升至王座法院(King’s Bench)的首席法官。他在各种法律职务中不折不挠,抵抗政治权威和国王对法律的侵犯。詹姆士一世试图将某些案件从普通法搬至教会法之下审理,柯克坚持说,国王没有足够权力来任意解释法律,从而引起极大愤怒。国王宣称,坚持国王在法律之下,无疑是叛国罪。柯克引用布拉克顿(Bracton)的话作答:“国王不应在人下,但应在上帝和法律之下(quod Rex non debet esse sub homine set sub deo et lege)。”⑫再加上其他的冒犯,柯克最终被解除一切法律职位,转而加入议会,成为反皇派领袖。
宗教作为集体行动的基础
跟法国、西班牙、匈牙利和俄罗斯不同,英国对专制权力的抵抗也涂上宗教色彩,大大加强了议会阵营的团结。第一任斯图亚特国王詹姆士一世,其母亲是被处决的玛丽·都铎(Mary Tudor),即苏格兰女王玛丽一世;其儿子查理一世(Charles I)娶法国路易十三的妹妹亨利埃塔·玛丽亚(Henrietta Maria)为妻。父子都表示相信新教,但常被怀疑对天主教抱有同情。大主教劳德(Laud)试图使英国国教向天主教靠拢,更加重视仪式,为此深受清教徒(Puritan)的憎恨。早期斯图亚特的专制主义教条和王权神授,与法国和西班牙的天主教君主的观点遥相呼应。很多新教徒从中看到国际天主教意欲剥夺英国人天生权利的大阴谋。1641 年爱尔兰的天主教叛乱仿佛就在家门口。新教徒移民遭受暴行的报告,似乎确认了很多英国人对国际天主教扩张的最坏担心。其中还真有一定的道理。西班牙国王在 16 世纪末派来无敌舰队(Armada),并投入八十年战争,以征服新教徒的荷兰联合省。法国的路易十四在 17 世纪末接过这项任务,出兵侵犯荷兰,他的秘密同情者就是英国最后一位天主教国王詹姆士二世。
有关英国内战的浩瀚史籍总有周期的修正。它不断学术性地改变对战争动机的理解,以跟上流行的思想风尚,以致有些历史学家对取得共识放弃希望。⑬ 20 世纪的很多解释,淡化了战争参与者的宗教动机,并将宗教意识视作阶级或局部的经济利益的面罩。事实上这段时期的宗教和阶级,其间互动非常复杂,很难厘清宗教和政治的效忠对象。有站在议会一边的国教徒,也有作为保皇派的新教徒。很多高层国教人士认为,与天主教会相比,像公理会(Congregationist)和贵格会(Quaker)那样的非国教徒,对道德秩序构成更可怕的威胁。⑭显然,较激进的新教流派变成了社会动员和经济进步的载体,并为抗议和团结提供机会,而传统的等级制的宗教渠道是无能为力的。
另一方面,即使有人主张冲突的主要原因不在宗教,但宗教在动员政治参与者和扩大集体行动范围上,仍然发挥重大作用。这在议会阵营,以及议会创建的新模范军(New Model Army)中,尤其如此。由于很多军官的宗教信念,随着时间的推移,新模范军变成反皇派激进主义的大温床。光荣革命期间,议会愿意接受奥兰治公国的威廉,以取代英国的合法君主詹姆士二世,就是因为前者是新教徒,后者是天主教徒。不然,真不好解说。
所以,英国地方上的自治团体、深植人心的法律、产权不可侵犯的信念、君主政体涉嫌参与国际的天主教阴谋,这一切都有助于议会阵营的精诚团结。
自由城市和资产阶级
现代传统智慧认为,如果没有强大中产阶级的存在,民主就不会出现。他们是有产阶级,既不是精英,也不是乡村穷人。这个概念起源于英国的政治发展,与其他任何欧洲国家(可能的例外是荷兰)相比,英国看到更多城市和城市资产阶级的早期涌现。城市中产阶级在议会中扮演主要角色,在内战和光荣革命之前,就已获得经济和政治的实质性力量。在权力的三角比赛中,它是抗衡领主和国王的大砝码。城市资产阶级的兴起,是更为广泛的西欧变迁的组成部分,包括低地国家、意大利北部和日耳曼北部的汉萨同盟(Hanseatic)港口城市。详细描述这一重要现象的有卡尔·马克思、马克斯·韦伯、亨利·皮朗(Henri Pirenne)。⑮马克思把“资产阶级的兴起”当作他现代化理论的中心命题,成为社会发展过程中必不可少的阶段。
我们在第 25 章中看到,自由城市的存在促成了西欧农奴的解放。对英国政治发展和议会获胜来说,强大且凝聚的资产阶级是非常重要的。但资产阶级在英国和西欧历史上所扮演的角色,在很多方面却是异乎寻常的。它是特殊境遇的后果,其他欧洲国家只是没遇上如此境遇而已。尤其是在易北河以东,那里只有很少独立自治的商业城市,遵照自己的法律,受自己民兵的保护。那些城市更像中国的,只是地方领主控制的行政中心,碰巧也充任商业中心。马克思的巨大影响促使好几代学生,继续把“资产阶级的兴起”看作经济现代化的伴随物,无须作出进一步解释,认定该阶级的政治力量来自其经济力量。⑯
早于马克思几乎七十五年,亚当·斯密在《国富论》中就资产阶级的起源,提供了更为周详、更具说服力的解释。他认为,在资产阶级的兴起当中,政治既是原因又是结果。斯密在第一卷第三章的篇首提出,他所谓的“富裕”(opulence),即经济增长,会有自然的升级,始于农业生产效率的改善,导致更多国内的城乡贸易,到最后才是日益增加的国际贸易。他注意到,欧洲现代国家所经历的次序恰恰相反:国际贸易发展在国内贸易之前,前者兴旺起来之后,强大男爵和地主的政治霸权才被打破。⑰
在斯密看来,造成这奇特次序有好几条原因。第一,罗马帝国崩溃后的大部分土地都在强大男爵手中,他们宁愿保住自己的政治权力,也不愿追求财产回报的最大化。所以,他们创建长子继承制和其他限制性的规则,以防地产的流失。此外,他们又将农民贬为农奴或奴隶;斯密认为,农奴或奴隶既不愿卖力干活,又不愿投资于土地。不愿追求回报最大化的另一原因,是缺乏以盈余去购买的消费品。在欧洲的黑暗时代,贸易不存在。因此,有钱有势者没有其他选择,只得与大批侍从共享盈余。⑱
斯密又注意到,出现于中世纪的城市,其最初居民是“商人和工匠”。他们属于低级阶层,甚至处于奴役地位,但是他们逃离了领主的控制,在城市找到庇护。久而久之,国王授予特权,让他们可以自由嫁女(编按:指无需领主同意而自主决定),组织自己的民兵,最终作为集团实体而享有自己的法律。这就是资产阶级的起源,虽然亚当·斯密没有使用如此字眼。不同于马克思,斯密提到独立城市的兴起必须有重要的政治前提:
领主鄙视市民,认为他们属于不同层次,只是被解放的农奴,几乎不是自己的同类。市民的富裕,常常使领主愤怒,一有机会就掠夺欺凌,不稍宽恕。市民自然也既嫉恨、又畏惧领主。恰好,国王也嫉恨和畏惧领主。国王虽可能亦会鄙视市民,但却没有理由去嫉恨和畏惧他们。所以,相互利益促使他们支持国王,又促使国王支持他们来反对领主。⑲
斯密接着说,这就是国王将独立宪章和法律赋予城市的原委,允许他们在国王与领主的斗争中成为一枚平衡砝码。
城市和资产阶级的形成,与马克思所相信的相悖,不只是经济增长和技术变化的结果。刚开始,他们非常软弱,从属于强大的领主,除非获得政治保护。这就是在波兰、匈牙利、俄罗斯和易北河以东其他土地上所发生的。那里,政治力量的不同配置使君主变得软弱,或促使君主与贵族的派别结盟,以反对市民利益。由于这个原因,东欧从来没有强大独立的资产阶级。技术上先进的资本主义市场,其引进者不是市民,而是进步地主,或国家本身,因此无法达到相似的繁荣程度。
基于城市的资本主义市场经济一旦出现,我们便离开古老的马尔萨斯式世界,开始进入现代经济制度,生产效率的增长变成家常便饭。此时,日益富有的资产阶级,越来越能颠覆旧式地主秩序的权力,政治发展的条件因此而发生变化。斯密表明,旧精英受财富的诱惑而放弃自己的政治权力——钻石扣环,“更适合于作孩子的玩具,而不应是大人的认真追求”——旧农业经济是无法创造这种财富的。⑳因此而开始了政治发展的现代制度:政治变化取决于经济和社会的变化。但一开始,资产阶级的兴起要有政治前提——市民和国王都憎恨领主。这个条件不存在的地方,如东欧的大部,就没有资产阶级的出现。
征税斗争
自 13 世纪以来,英国议会开始定期开会,比法国、西班牙和俄罗斯更为频繁。如上所述,它们的原始功能是司法,但久而久之,开始扮演更广阔的政治角色,成为国王的联合统治者。在批准税赋上,议会作用尤其重要,因为议会包括全国大多数地主,其资产和收入是国家征税的基础。到 14 世纪和 15 世纪,下议院与英国君主密切合作,以剔除不够格或腐败的官员,并定期监督议会拨款的具体花费。㉑图 5 显示的是内战前夕的 1641 年的英国社会力量。
查理一世在 1629 年解散议会,开始了十一年的“亲政”,试图以议会为代价来扩展国家权力。这导致查理一世与议会对手在好多问题上发生争执,有的已在前面篇幅介绍过。议会中很多人不喜欢大主教劳德的专制国教,怀疑查理一世同情天主教,因为他有兴趣与法国和西班牙建立外交关系。宗教问题和保卫法治互相交融,星室法庭、高级专员公署(High Commission)、北方政务会(Council of the North)起诉反主教制(anti-Episcopal)的清教徒。清教徒传教士亚历山大·莱顿(Alexander Leighton),遭到星室法庭野蛮逮捕和残酷折磨,却得不到正当法律程序的保护,被认为是宗教和皇家权力肆无忌惮的滥用。
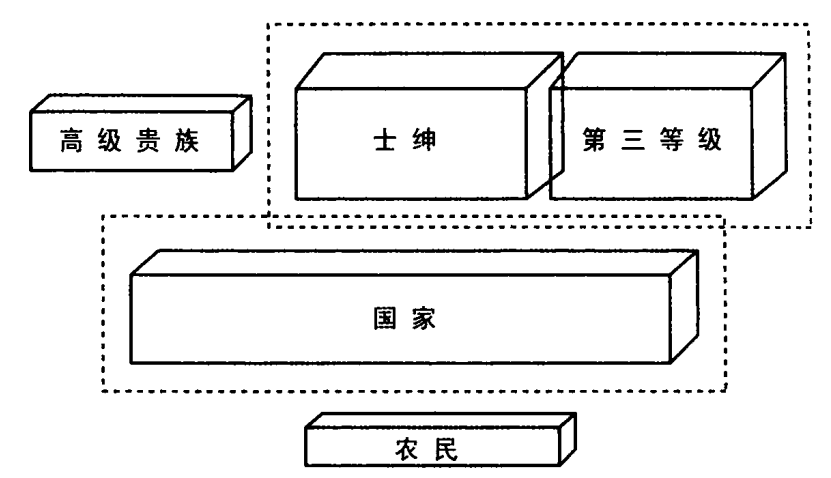
其时还有两大问题,一是没有议会批准、国王擅自增税的权利。国王提出新关税,向地主施以任意的罚金,重新引进蓄意避开禁令的垄断专利,在和平时期为重整海军而征收“船舶筹款”。㉒英国税务制度的发展不同于法国,英国的贵族和士绅未以法国方式购买特权和免税,税收负担的最大部分都落到了议会所代表的富人头上。可能与紧密的地方团结有关,富人阶层没有与国王共谋,将税收负担推向农民、工匠和新近致富的中产阶级,反而认为自己与议会的权力和特权休戚相关。
第二个问题涉及政治腐败。与法国和西班牙一样,英国也躲不过家族化和买卖公职的做法。从都铎时期开始,皇家公职的获得愈益依靠政治赞助,晋升不是选贤与能,而是以各种荫庇关系圈子的圈内人资格为准。㉓公职待价而沽,又变成世袭财产。到斯图亚特王朝早期,法国包税(关税)和内部财政(向国家官员借款)的做法获得引进。国王建立皇家调查委员会,就像法国的司法堂,以私人腐败的借口敲诈富有官员。㉔
1641 年爆发的内战持续十年,最后在 1649 年,以议会的胜利和查理一世被砍头而告终。但国王和议会的长期斗争,其最终解决并不全然依靠武力,虽然暴力和暴力的威胁仍是重要的决定因素。㉕胜利的议会派因处决国王而抹黑了自己声誉;在克伦威尔(Oliver Cromwell)护国公时期,又因追求愈益激进的政策而削弱了自己的政治基础。所以,查理一世的儿子在 1660 年成功复辟,登位为查理二世,反而带来一种解脱。二十年的强烈政治冲突之后,国家得以返回常态。
复辟确实解决了当初引起内战的腐败问题。议会在内战和护国公时期推行很多政府改革,譬如建立严密的现代新模范军和清洗腐败的皇家官员。但查理二世的上台,又带回很多早期斯图亚特的腐败做法,包括出售公职和赞助任命。但是,多种因素聚合起来,在英国政府中建立了改革同盟,最终打退这些倒行逆施。
首先是第二次荷兰战争(1665—1667)爆发,加上瘟疫突发和伦敦大火,导致国防严重衰弱,以致荷兰船队溯泰晤士河而上,烧毁英国海军船坞。路易十四治下的法国也取得进步,以咄咄逼人的外交政策威胁欧洲大陆的均势。显然,军费必须上涨。第二是查理二世希望量入为出,以避免向议会请求特别征税。第三是政府中出现了一批才华横溢的精明改革家,包括乔治·唐宁(George Downing)爵士和喜记日记的塞缪尔·佩皮斯(Samuel Pepys)。他们关心日益增长的外国威胁,认识到财政制度和行政管理必须改革才能获得高效。㉖最后,告别内战和护国公时期的议会,对政府的浪费和腐败深感怀疑,其时政府将税收用于非公共的开支。
不同压力的汇合允许唐宁组织的第二财政委员会(Second Treasury Commission)推荐和实施重要的改革,使英国公共行政管理更为现代化,进一步脱离家族化。它取消从都铎时期起便是腐败温床的国库(exchequer)的权力,移交给总管所有政府开支的新财政部。它向公众发行遵守公共债券市场纪律的新债券(Treasury order),以取代内部财政。最后,它将私人拥有的公职改成“悉听尊便”(at pleasure)的职位,并取消新的公职出售。㉗
1667 年后发生的改革努力沉重打击了家族化实践,确保英国在管理公共资金上比法国或西班牙更为有效。反对腐败政府的斗争从来不是一蹴而就,唐宁在 17 世纪 60 年代发起的很多改革,其完全实施尚要等到 18 世纪早期。这些努力也没有排除后续稽查的需要,因为假以时日,家族制总是试图卷土重来。
17 世纪晚期确实提供了扭转家族化的重要模式,对今天的反腐努力仍有意义。促使晚期斯图亚特王朝改革的所有因素依然重要:外部环境的压力迫使政府改善效率;首席执行官如果没有发挥带头作用,至少不拖后腿;政府内有人倡导改革,并得到足够的政治支持来付诸实行;最后,来自纳税人的强大政治压力,他们不愿看到浪费。
国际机构最近作出的很多反腐努力,比如世界银行或英国国际发展部,但却功亏一篑,就是因为上述因素之一的缺席。现代世界的问题在于,腐败政府经常无需向自己公民谋求税收,像查理二世所作的,因此没有议会或公民社会来监督它们的开支。它们的收入或者来自自然资源,或者来自并不要求财政负责制的国际捐赠人。亨廷顿建议,如果英国议会的呐喊是“无代表即不纳税”,今天口号应该是“不纳税即无代表”,因为最能激励政治参与的乃是后者。㉘
光荣革命
国王与议会争斗的结果是 1688—1689 年的光荣革命,詹姆士二世被迫退位。奥兰治公国的威廉从荷兰赶来,登基为国王威廉三世。直接原因是天主教徒的詹姆士二世试图扩军,并配以天主教军官。这即刻引起怀疑,他是否打算利用军队实施专制,并与法国和其他天主教势力结成同盟。更大原因则与议会当初反对国王导致内战的原因相同:合法性最终应基于被统治者的同意,得不到同意,国王无权强加于人。危机中达成的和解,涉及宪法、宗教、财政、军事等重要方面。在宪法上,它建立了没有议会同意国王不得建军的原则;议会还通过议案,罗列国家不得侵犯的国民权利。在财政上,它确立没有议会同意国王不得征新税的原则。在宗教上,它禁止天主教徒成为国王或王后,还添上增加异见新教徒权利的容忍议案(但排除天主教徒、犹太教徒和索齐尼派教徒)。㉙最后,它允许政府发行更多债券,使国家机构的大大扩展成为可能。议会主权的原则要在数年后才得到最后确认,光荣革命不愧为现代民主发展的主要分水岭。㉚
光荣革命导致了政治合法性的思想大改变。作为这些事件的评论家和参与者,哲学家约翰·洛克扩充了霍布斯的观点,即国家源于为保障天赋权利而签署的社会契约。㉛其《政府论》上篇攻击罗伯特·菲尔麦(Robert Filmer)爵士为君主政体辩护的君权神授;其《政府论》下篇力辩,与霍布斯相悖,侵犯臣民天赋权利的暴君可被撤换。这些原则使用普世性的论述,对 1689 年的宪政和解至关重要。光荣革命不是某个统治者或一群精英从他人手中夺得国家和租金,而是定出如何选择后续统治者的原则。从洛克的《政府论》下篇,到美国革命和美国创始人的宪法理论,其间距离很短。尽管现代民主有复杂的方方面面,但 1688—1689 年的事件,牢固建立了政府合法性来自被统治者的同意的基本原则。
光荣革命使政治负责制和代议政府的原则制度化,但还没引进民主。此时的英国议会,只由很小比例的人口选出。出席议会的有高级阶层、议员和士绅。后者是英国最重要的政治阶级,根据彼得·拉斯莱特(Peter Laslett),它代表了大约总人口的 4% 到 5%。㉜更为广泛的民众参与地方统治,或参加陪审团,或协助百户区和县政府的工作,包括大部分条件较好的自耕农(yeoman)。如把这个团体也包括在内,政治参与者会接近男性成人总人口的 20%。㉝我们今天理解的民主——无论性别、种族、社会地位,所有成年人都享有选举权——要到 20 世纪的英国或美国,方才得到实施。跟美国独立宣言一样,光荣革命建立了被统治者同意的原则,让后人再去拓宽政治意义中的“人民”的范围。
有些人认为,光荣革命的重要性在于它标志了英国安全产权的开始,其实非也。㉞数世纪之前,健全产权即已建立。包括女子在内的个人早在 13 世纪就行使买卖财产的权利(参看第 14 章)。普通法、国王法庭、县法庭和百户法庭,允许非精英地主在领主司法范围之外,提出产权争执的诉讼。到 17 世纪晚期,强大的资本主义经济,参与反斯图亚特专制的中产阶级,都已出现。光荣革命的成功,与其说是强大可靠产权的原因,倒不如说是它的结果。英国有产阶层觉得有重要东西需要保护。
光荣革命也未给新近壮大的纳税人减税的借口,如经济学家曼瑟尔·奥尔森所提示的。㉟恰恰相反,政府开销作为国民生产总值的百分比,从 1689—1697 年的 11%,涨至 1741—1748 年的 17%,再涨至 1778—1783 年的将近 24%。㊱在 18 世纪的高峰时期,英国征税高达 30%。
光荣革命的重大成就之一是使征税合法,从此以后,征税全凭同意。民主政体的公众并不一定反对高税,只要知道这是为了重要的公共目标,比如国防。他们所不喜欢的是非法征税、税款被浪费或掉进贪官污吏的荷包。光荣革命之后,英国投入两场与路易十四法国的昂贵战争,即九年战争(1689—1697)和西班牙继位战争(1702—1713)。二十年几乎连续不断的战争,证明是非常昂贵的。从 1688 年到 1697 年,英国舰艇的数量几乎翻了一番。纳税人愿意支持这些及后来的战争,因为他们在战争得失上得到咨询,被要求批准新的税收。不用多作解说,英国较高的征税并没有遏制资本主义革命。㊲
与专制法国的对照是很醒目的。法国没有接受同意的原则,征税必须依赖强力。政府在同一时期从没能征收超过其国民生产总值的 12% 到 15%,真正到手的往往更低。法国社会中最负担得起的精英却在购买免税和特权,这意味着税收负担落到社会最弱成员的身上。所以,在路易十四过世的 1715 年,人口几乎是英国四倍的法国发现自己已经破产。
光荣革命和随之发生的财政和银行改革,如 1694 年建立的英国银行,确实使公共财政经历了革命性的变更。它们允许政府在透明的公共债务市场上借贷资金,而法国或西班牙是无法企及的。因此,英国政府借贷在 18 世纪激增,使得国家愈加壮大。
美国革命和法国大革命
本卷对政治发展的介绍到此结束,时值 18 世纪末,美国革命和法国大革命的前夕。在此停下有逻辑上的原因。黑格尔的伟大注释者亚历山大·科耶夫(Alexandre Kojève,出生于俄罗斯,后来长住法国)认为,众所周知的历史终结于 1806 年,其时,拿破仑在耶拿和奥尔斯塔特击败普鲁士君主政体,将自由和平等的原则带到黑格尔的欧洲。科耶夫以他通常的讽刺和顽皮说明,1806 年以来发生的一切,包括间杂世界大战和革命的 20 世纪狂飙突进(sturm und drang),只是在填平历史所留下的坑坑洼洼。也就是说,现代政府的基本原则在耶拿战役时已获建立,后续任务不是发现新的原则和更高级的政治秩序,而是将之推至世界上越来越多的地区。㊳
我相信,科耶夫的声明仍值得认真考虑。现代政治秩序的三个组件——强大且有能力的国家、国家从属于法治、政府对所有公民负责——已在 18 世纪末世界上的某地获得确立。中国很早就开发了强大国家;法治存在于印度、中东、欧洲;负责制政府首次出现于英国。耶拿战役之后的政治发展,只涉及这些制度在全世界的复制,而没有看到全新制度的补充。20 世纪的共产主义曾有如此的雄心壮志,到了 21 世纪,却又在世界舞台上几近消失。
英国是三大组件聚合在一起的第一个大国。这三者互相之间高度倚靠。没有强大的早期国家,就没有法治,以及对合法产权的广泛认识。没有健全的法治和合法产权,平民绝不可能群起奋争,将负责制强加给英国君主政体。没有负责制的原则,英国绝不可能在法国大革命时成为强大国家。 其他欧洲国家,包括荷兰、丹麦和瑞典,也在 19 世纪建立国家、法治和负责制的整套制度。它们所走的途径与英国有实质上的不同。但要承认,整套制度一旦到位,它所创建的国家如此强大,如此合法,对经济增长如此友善,以至成为推向全世界的模式。㊴在缺乏英国式历史和社会条件的国家,这个模式将有怎样的遭遇,那将是第 2 卷的主题。
第 28 章 负责制或专制主义?
前述案例的互相比较;通向代议政府的英国路径不是唯一;达到丹麦;历史讨论与今日的民主斗争息息相关
我们现已介绍了五个欧洲案例,在负责制和代议制度方面,引出四个不同结果。法国和西班牙创造了弱的专制主义,但没有建立议会负责制的原则。两个国家分割出卖自己的功能给众多精英,精英以特权和免税保护自己——但不包括其余社会群体——避开国家的任意权力。俄罗斯建立了更为彻底的中国式专制主义,其君主政体将精英征入国家服务,予以掌控。在匈牙利,强大凝聚的精英在君主权力上实施宪政制衡,从而建立起负责制的原则。但这制衡太过强大,以致阻碍了国家的有效运作。最后,只有在英国,强大的议会将负责制原则强加于国王,但并没有破坏强大和统一的主权政府。问题在于,为何会有如此不同的结果?
可用一个很简单的公式来解释这些差异,其与我们所介绍的农业社会中四大政治参与者的均势有关。它们是以国王为代表的国家本身、高级贵族、士绅以及我所谓的第三等级。这种四分法过于简略,但对结果的理解大有裨益。
欧洲有些贵族家族先发制人,取得优势,而变得强盛起来——法国的卡佩家族、匈牙利的阿尔帕德王朝、俄罗斯的留里克王朝、征服后的诺曼王朝——从而出现国家。它们的兴起归功于有利地理、卓越领导、组织能力和掌控合法性的能力。合法性可能是统治者最初优势的来源,如率领马扎尔人(Magyars)皈依基督教的伊斯特万。有人以赫赫武功征服军阀对手,给社会带来和平和安全,合法性也可能接踵而至。
高级贵族可说是遗留下来的军阀,拥有自己领土、大批侍从和资源。这个群体有效治理自己的领土,可传给后裔,也可交换成其他资产。
士绅是低级精英,虽有社会地位,但不一定拥有重要的土地或资源。他们的人数远远超过贵族,明显从属于贵族。
第三等级包含工匠、商人、解放了的农奴,以及不受庄园经济和封建法律管辖的城镇居民。
除了这四个群体,还有占人口大多数的农民。然而,农民还不是重要的政治参与者。到 18 世纪,他们才在北欧某些地区参与政治。四下分散、贫困和缺乏教育的农民,很难完成重大的集体行动。中国、土耳其和法国的农业社会,农民起义同期性爆发,最终都被镇压,经常伴随可怕的野蛮和残忍。那些反抗影响了其他参与者的行为和计算,例如,国家在考虑增加农业税时会特别小心。在其他时刻,农民起义可帮助推翻中国皇朝。但农民很难采取集团行动,以迫使整个制度实施关心农民利益的长期改革。
这五个群体的交叉关系在图 1 中得到说明(参看第 22 章)。除农民外,这些社会群体都组织起来(只在程度上有深有浅),可以成为政治参与者,为夺得权力而斗争。国家尝试扩充自己的统治。国家之外的群体试图保护和扩充现有特权,或反对国家,或互相争斗。这些斗争的结果多半取决于主要参与者的集体行动,甚至国家本身也需要精诚团结。王朝的内部分裂、组织故障、侍从不再相信王室的合法性、国王没能孕育继位者,都有可能造成国家的软弱。此外,这些不同群体可以组成各式同盟——国王和士绅之间、国王和第三等级之间、高级贵族和士绅之间、士绅和第三等级之间,等等。
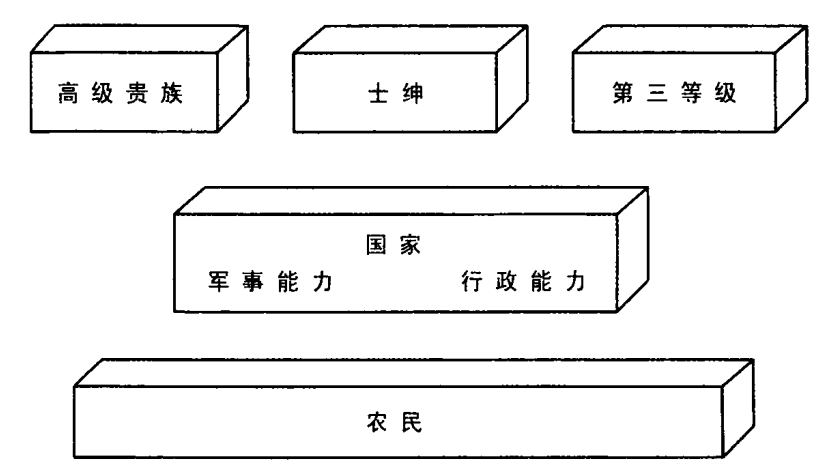
出现专制主义的案例中,无论是强大的还是软弱的,抵抗国家的群体不可避免地遇上了集体行动的故障(参看图 6)。哪里有负责制,哪里的国家相对弱于其他政治群体。议会政府出现的地方,凝聚的国家和组织良好、善于自卫的社会之间产生了相对均势。
弱的专制主义
我们现在可以总结早先章节所描述的各种结果。
相对软弱的国家遇上组织良好的社会,但前者仍得以掌控后者,这就是法国和西班牙,遂出现弱的专制主义。在这两个案例中,国家的权力基础依靠有限的领土,包含皇家领地和国家直接征税地区——对法国君主政体来说,即巴黎周围的财政区省(编按:原文误为三级会议省[pays d’états],据前文第 23 章改正);对西班牙哈布斯堡王朝来说,即卡斯提尔区域。它们都想通过拉拢收买、王朝阴谋和直接征服来取得更多领土,但西欧的地理,以及 16 世纪晚期和 17 世纪早期的军事技术,尚不允许快速的武力扩张——应该还记得,星状要塞使围攻战争变得既昂贵又必不可少——法国和西班牙的君主很快发现,自己因军事开支和帝国扩张而深陷财政危机。
在这两个案例中,国家之外还有强大的地方参与者,竭力抵制中央集权。他们包括拥有土地和资源的古老血缘贵族、广大的士绅阶层、城市资产阶级,已经组成正式的等级——法国的高等法院和西班牙的议会。法国和西班牙国家一步步收买这些群体,开始时好像不是国家建设的战略计划,只是防止破产的绝望革新。最初,法国在财政区省以免税和特权来购买地方精英的忠诚。它在 1557 年对“大借款”赖债不还,引发破产。这之后,它开始向富有个人出售公职,到 17 世纪早期又摇身变为世袭财产。公职的出售和再出售,一直持续到世纪末的路易十四年代。意大利和低地国家的持久王朝战争,使西班牙国家早早陷入破产。来自新世界的收入使之维持到 16 世纪末。到 17 世纪,它也只好诉诸拍卖国家的部分职能。
法国和西班牙君主的集权能力,受到两国既存法治的严格限制,他们觉得必须尊重封建权利和臣民的特权。但他们试图抓住每一次机会,以扩充征税和征兵的权力,一有可能就想方设法扭曲、违反或规避有关法律。他们鼓励知识分子传播专制和主权的教条,以支持自己是法律最终来源的宣称,但没有设法废除或忽略法律。到最后,他们仍受规范化的遏制,无法从事中国皇帝式的随心所欲,像血腥清洗贵族对手的武则天,或像任意没收豪族土地的明朝开国皇帝朱元璋。
对精英的一步步收买,意味着一再扩展寻租联合体,先是传统的贵族精英,再是新动员起来的社会参与者,如城市资产阶级。更确切地说,与其联合起来保护自己阶层的利益,这些精英宁愿以政治权力来交换社会地位和部分国家职能——不是议会的代表权,而是国家征税权的分享。用托克维尔的话说,自由被理解为一种特权,而不是真正的自治。这导致弱的专制主义,一方面,国家权力不受正式宪法的约束,另一方面,它又将自己前途抵押给了自己很难掌控的大批强势个人。
对法国和西班牙来说,国家的软弱最终证明是致命的。因为国家建设以精英免税为基础,税收负担都落到农民和普通商贩的头上。两个国家都无法征集足够收入,以满足统治者的帝国野心。法国竞争不过更小的英国,后者的税收基础因议会负责制的原则而获得保证。西班牙进入持续一世纪的军事和经济的衰退。它们的国家都丧失了合法性,因为其组成方式本身就是腐败的,法国改革的失败为大革命铺平道路。
强大的专制主义
俄罗斯建立了更接近中国的强大专制主义国家。只要将它的发展与法国或西班牙作一对比,个中原因显而易见,其间重大差异至少有五项。
第一,俄罗斯的地理——广阔大草原,只有很少自然障碍来应对基于骑兵的军队——使之易受来自西南、东南和西北的入侵,且经常是同时发生的。军事动员因此而变得非常重要。这还意味着,与对手相比,先发制人的军阀享有规模上的极大优势。莫斯科国家的权力基于对服役贵族——相当于士绅——的军事招聘。它能这样做,因为它仍是边界不定的边境国家。跟奥斯曼帝国西帕希骑士的情形一样,该阶层成员所获的奖励就是新土地,这些骑兵变成了国王的直接受养人。(最相似的西欧做法就是西班牙国王,将新大陆的托管权作为服务的报酬赠与征服者,导致了同样的政治等级制度。)莫斯科公国凭借对鞑靼的早期军事成功而获得先发制人的优势,因此享有比其他封地领主更多的合法性。
第二,从卸下鞑靼轭到莫斯科投入国家建设,其间只有很短时期。封建主义在西欧扎根八百年,孕育了骄傲的血缘贵族,坚守在散布四野的险固城堡。相比之下,俄罗斯的封地时期仅持续两个世纪,贵族成员组织松弛,根本无法抵制中央君主的权力,也没有城堡可住。与西欧相比,他们以及像诺夫哥罗德那样的独立城市,较少受到地理上的保护。
第三,俄罗斯没有可与西欧媲美的法治传统。指派俄罗斯牧首的拜占庭东正教,本身没有经历类似叙任权斗争的冲突,始终是政教合一,直到君士坦丁堡的失陷。拜占庭帝国的法律也没变成综合法典,受西方那样法律专业自治团体的保护。俄罗斯东正教是拜占庭教会的精神继承人,虽然时有偏离莫斯科统治者的政治独立,但也从国家赞助那里收获重大好处。天主教会可在分裂的西欧政治舞台中合纵连横,俄罗斯教会没有选择,只好去莫斯科,通常成为国家的顺从拥护者。没有独立的教会权威来监看一套教会法规,这意味着,接受训练的法律专家没有属于自己的机构来培养集团身份。教会官僚担任早期西欧国家的行政官员,在俄罗斯,管理国家机构的是军官和家族人选(经常是同一人)。最后,对很多俄罗斯人来说,统治者的榜样不是依法执政的君主,而是纯粹掠夺的蒙古征服者。
第四,地理环境使农奴所有者结成卡特尔成为必需,将贵族和士绅的利益与君主政体的利益紧紧绑在一起。因为没有地理界限,要维持像农奴制那样的制度,全靠农奴主在处罚和归还逃奴上的严格自律。沙皇只要支持对农奴实施越来越紧的限制,就可把精英拴在国家这一边。相比之下,西欧的自由城市是庇护所,逃离领主和庄园经济的农奴,为追求自由而来投奔城市。在俄罗斯,城市充任功能上的边境——最终都被封闭。明显不同于俄罗斯君主和其他东欧统治者,西欧的国王发现,自由城市在反对强势领主的斗争中于己有利,因此予以保护。
最后,有些思想在俄罗斯的渗透,达不到在西方国家那样的程度。首先是法治,再延伸到源于宗教改革和启蒙运动的整套思想。丹麦王太后索菲亚·玛德莲娜(Sophie Magdalene)在皇家领地上释放农奴时,曾是伏尔泰朋友的叶卡捷琳娜大帝,却在对俄罗斯农奴的行动自由实施更为严格的限制。当然,很多启蒙运动的思想被推行现代化的俄罗斯君主所采用,像彼得大帝。三代之后,沙皇亚历山大二世方才解放农奴。与欧洲其他部分相比,现代思想对俄罗斯的影响更为缓慢、更为虚弱。
英国的结果为何不像匈牙利?
以这些未能抵抗专制的国家为背景,英国的成果便显得愈加惊人。英国主要社会群体为保护自己的权利而反对国王,所显示出的团结胜过任何其他地方。英国议会包括从大贵族到自耕农的全部有产阶级的代表。其中两个群体特别重要,那就是士绅和第三等级。前者的阶层没有被招募进入国家服务,像俄罗斯那样;后者基本上不愿以政治权利来交换爵位和个人特权,如法国那样。法国、西班牙、俄罗斯的君主政体,向精英中的个人兜售官爵,从而破坏精英之间的团结。俄罗斯的门第选官法,其目的很像法国和西班牙的卖官鬻爵。英国君主也尝试像出售公职那样的手段,但议会仍是凝聚的机构,其原因早已提到——即对地方政府、普通法和宗教的普遍认可。
这还不足以解释英国议会为何如此强大,以致君主政体被迫接受宪法。匈牙利议会中的贵族也很强大,也组织良好。像在兰尼米德的英国男爵,较低层次的匈牙利贵族在 13 世纪强迫君主接受宪法妥协,即金玺诏书,在后续年份中又死死看住国家。①在马蒂亚斯·匈雅提于 1490 年去世后,贵族阶层扭转君主政体在前一代作出的中央集权改革,夺回权力。
但匈牙利贵族阶层没有运用权力来加强整体国家能力。更确切地说,他们以国防能力为代价给自己减税,保护自己的狭隘特权。相比之下,源自 1688—1689 年光荣革命的宪政和解,大大加强了英国的国家能力,以至它在未来一世纪中成为欧洲的主要强国。那么,我们要问的是,既然英国议会已经强大到能够遏制掠夺性的君主,但议会本身为何没有发展成寻租联合体,没有像匈牙利议会一样作茧自缚。
英国负责制政府没有退化成贪婪的寡头政治,至少有两条原因。首先,英国的社会结构不同于匈牙利。英国议会中的团体是寡头政治,但与匈牙利相比,他们底下的社会更为流动,向非精英开放的程度更高。在匈牙利,士绅被吸收到狭窄的贵族阶层;在英国,他们代表一个庞大且凝聚的社会群体,在某些方面甚至比贵族还要强大。不像匈牙利,英国拥有基层政治参与的传统,体现在百户法庭、县法庭和其他地方治理机构。英国领主习惯于出席会议,与自己的属臣和佃户平等相处,讨论决定涉及共同利益的问题。此外,匈牙利没有英国那样的自耕农。自耕农是相对富裕的农民,拥有自己的土地,参与地方上的政治生活。匈牙利城市受到贵族阶层的严格控制,不能像英国那样衍生出富庶和强大的资产阶级。
其次,尽管英国有个人自由的传统,但它的中央国家既强大,又在社会上获得大致的好评。它是发展出统一司法制度的首批国家之一,保护产权,为应付欧洲大陆的各式敌人而建起实质性的海军。1649 年查理一世被送上断头台,之后,英国又试验了共和政府和克伦威尔的护国公体制,结果都不如人意。甚至在议会拥护者的眼中,弑君本身似乎都是不正义的非法行为。英国内战见证了同样的愈趋激进,像法国、布尔什维克和中国革命后来所经历的一样。更为激进的反皇派,像平均派(leveller)和掘地派(digger),所追求的似乎不只是政治负责制,而是更为广泛的社会革命,从而吓坏了议会所代表的有产阶级。所以,随着查理二世的登基,君主政体在 1660 年得以复辟,大家都松了一口气。②复辟之后,政治负责制的问题在天主教徒詹姆士二世的治下重新泛起,其阴谋诡计招致议会的怀疑和反对,最终引致光荣革命。但这一次,没人再想废除君主政体或国家,只想要一位对自己负责的国王,那就是奥兰治的威廉。
这再次证明思想是非常重要的。到 17 世纪晚期,像霍布斯和洛克那样的思想家,摆脱了基于阶级和等级的封建秩序的概念,转而赞成国家和公民之间的社会契约。霍布斯在《利维坦》中认为,就激情和彼此施暴的能耐而言,人与人在根本上都是平等的;此外,他们还享有天赋权利。洛克接受这些前提,并抨击不经被统治者同意也可有合法统治的主张。只要依照同意原则,就可以推翻国王。这些早期自由主义者认为,权利是抽象和普遍的,任何强人不得合法剥夺。但在这些思想传到匈牙利之前,匈牙利早已向土耳其和奥地利屈服称臣了。
从上述比较中可得出一个简单的教训。政治自由——即社会自我统治的能力——不但要看社会能否动员起来,以对抗中央集权,并将宪法约束强加于国家;还要看国家是否足够强大,能在必要时采取行动。负责制不是只从国家流向社会的单行道。如果政府不能采取团结行动,没有广泛接受的共同目标,就无法奠定政治自由的真正基石。明显不同于马蒂亚斯·匈雅提死后的匈牙利,1689 年之后的英国仍然强大而团结。议会愿意向自身征税,为 18 世纪的海外持久争斗作出牺牲。与没有制衡的政治制度相比,高度制衡的不一定会取得更大成功,因为政府需要定期采取坚决果断的行动。所以,负责制政治制度的稳定,全靠国家和社会之间的广泛均势。
达到丹麦
辉格史观的问题之一在于,它将英国的经历当作宪政民主制(constitutional democracy)兴起的范例。然而,欧洲其他国家走上不同路径,最后抵达与英国相同的目的地。我们开始解说冗长的政治发展史时,曾提出丹麦如何变成今日丹麦的问题——守法、民主、繁荣、执政清廉的政体,其政治腐败处于世界最低水平——我们需要时间来解释这个结果。
在 1500 年,还看不出丹麦(或斯堪的纳维亚其他国家)将会不同于中世纪晚期的其他欧洲社会。有些评论家尝试将今日的丹麦一直追溯到当初定居斯堪的纳维亚的维京人。③除了他们不骑马,驾长艇远行,很难想象这一掠夺部落,如何将自己从来自罗马帝国之后定居欧洲的日耳曼野蛮人中彻底区分出来。
丹麦的君主政体具有很古老的血统,从 13 世纪起变得相对软弱。国王被迫签署宪章,允诺向贵族议会征求咨询,向教会提供特权。④像欧洲的其他国家,丹麦的经济仍以庄园为基础。丹麦地处波罗的海的进口,邻近汉萨同盟的港口城市,使国际贸易成为其经济发展的重要因素。⑤卡尔马联盟(Kalmar Union)在 15 世纪中期曾短暂地统一大部分的斯堪的纳维亚。联盟解散后,丹麦仍是相当重要的多民族政权,控制着挪威、冰岛、说德语的石勒苏益格—荷尔斯泰因地区(Schleswig & Holstein),以及现是海湾对面的瑞典西部省份。
如果说有一个事件,促使丹麦和斯堪的纳维亚其他地区走上独特发展道路,那就是宗教改革。跟在欧洲其他地区一样,马丁·路德(Martin Luther)的思想证明是非常震撼人心的,催化了大众对天主教会的长期不满。在丹麦,短暂内战导致新教徒的胜利,以及 1536 年路德派丹麦国教的建立。⑥促成这个结果的,既有道德因素,也有物质因素,丹麦国王视之为攫取教会资产的良机。当时,教会拥有相当多的财富,大约占有丹麦土地的 30%。⑦
宗教改革在丹麦的持久政治影响是鼓励农民脱盲。路德教会坚信,普通老百姓要与上帝沟通,需要阅读圣经,或至少要读路德教的简易问答集(Lesser Catechism)。始于 16 世纪,路德教会在丹麦每一座村庄设立学校,让教士向农民传授基本的读写技能。结果在 18 世纪,丹麦农民(还有斯堪的纳维亚的其他地区)已成为教育程度相对较高、日益组织起来的社会阶层。⑧
当代社会的社会动员通常是经济发展的结果,这也是中世纪英国所走的道路。普通法的产权扩展,促使英国农民的最高层进化成政治上活跃的自耕农。相比之下,在前现代 16 世纪的丹麦,促进社会动员的是宗教。脱盲允许农民改善经济条件,还帮助他们互相交流,并作为政治行动者组织起来。到 19 世纪早期,斯堪的纳维亚和俄罗斯的乡村,彼此之间的悬殊是无法想象的,尽管两者的地理相近,气候相似。
跟英国的情形不同,这里的代议民主制并不来自组织良好、足以抵抗中央国家的封建残余机构(议会)。丹麦在 1660 年败于瑞典,遂建立了专制国家,其官僚机构变得愈益精明。⑨丹麦议会已被废除,没有基于社会等级的政治结构,可供君主前去要求增税。
从 1760 年到 1792 年,丹麦发生了重大的政治革命。开明君主开始逐渐废除农奴制(Stavnsbånd),先在皇家领地,再扩展到所有地主,并限制地主处罚下人的权利,譬如不能再将农民放在木马上鞭打。⑩农民仍然没有选举权,但可以拥有土地,并能在同等的基础上从商。⑪
丹麦君主将农民自由视作遏制贵族地主的良机,遂遭到了地主的顽强抵抗。他又可将获得自由的农民,直接征募进国家军队。思想也很重要。亚当·斯密的《国富论》出版于 1776 年,他认为,自耕农的生产效率将远远超过农奴。同样重要的是农民本身得到越来越多的教育和动员,充分利用自由经济的机会,投入到盈利较多的增值生意,例如食物加工。
使丹麦现代民主成为可能的第二个重大事件来自外国。到 18 世纪末,丹麦仍是欧洲中等的多民族政权。在 1814 年的拿破仑战争中失去挪威。19 世纪前几十年,法国大革命思想的传播促成复杂的政治后果。它刺激了基于阶级的两项需求,一项来自资产阶级和农民,跟政治参与有关;另一项来自说德语的少数民族,与国家认可有关。
普鲁士解决了第二项需求,通过一场短暂但决定性的战争,在 1864 年兼并了主要说德语的石勒苏益格—荷尔斯泰因公国。只过一个晚上,丹麦就变成基本上讲丹麦语的整齐划一的小国。它知道自己别无他法,只好接受小国寡民的处境。
丹麦的民主出现于 19 世纪后期,社会民主主义出现于 20 世纪早期,这就是它们的来龙去脉。教士兼教育家的葛隆维(N. F. S. Grundtvig)所激发的农民政治运动,最初只装扮成宗教复兴运动。它摆脱官方的路德教会,在全国各地大办学校。⑫等到立宪君主政体在 1848 年当政,农民运动和代表资产阶级的自由派开始要求直接的政治参与,并在翌年获得选举权。丹麦在 20 世纪成为福利国家,这已超越本卷的范围。当它最终来到时,并不完全依靠新兴的工人阶级,还需要农民阶级的帮助。在关键时刻,促使农民动员起来的不是经济增长,而是宗教。
民主和现代市场经济在丹麦的发展,比在英国经历了少得多的冲突和狂暴,更不用提相比法国、西班牙和德国了。为了到达现代丹麦,丹麦人确实与邻国打了好几仗,包括瑞典和普鲁士,也在 17 世纪和 19 世纪发生了激烈的国内冲突。但没有持久的内战,没有圈地运动,没有专制暴政,没有早期工业化所带来的赤贫,所留下的阶级斗争遗产非常薄弱。就丹麦的故事而言,思想是至关重要的,这不仅指路德教会和葛隆维的意识形态,而且还有 18 世纪和 19 世纪一系列丹麦君主所接受的关于权利和宪政的启蒙思想。
丹麦的民主兴起充满了历史的偶然性,不能在别处复制。丹麦抵达现代自由民主制的途径完全不同于英国,但最终都抵达非常相似的目的地。它们都发展了强大国家、法治和负责制政府。这似乎显示,“达到丹麦”可有多种途径。
第五部分 迈向政治发展理论
第 29 章 政治发展和政治衰败
政治的生物基础;政治秩序的进化机制;政治不同于经济;制度的定义;政治衰败的来源;国家、法治、负责制的相互关联;政治发展条件的历史演变
本卷提供的政治发展史是从前人类时代到法国和美国革命前夕,直到这时,真正的现代政治方才问世。此后,众多政治体出现,囊括现代政治制度的三大重要组件:国家、法治、负责制政府。
至此,有些读者可能会断定,我对政治发展的解读是历史决定论的。通过介绍各种政治制度复杂且背景独特的起源,我似乎在主张,类似的制度要在今日出现必须要有类似条件,各国因独特的历史背景已被锁定在各自单一的发展路径上。
这肯定是误解。能把优势带给社会的制度,总是被他人复制和改进;知识和制度的跨社会交汇,伴随着历史的始终。此外,本卷的历史故事,结束于工业革命前夕,而工业革命本身,又大大改变了政治发展的条件。这两点,将在最后一章得到详细描述。本书的第 2 卷,将描述和分析后马尔萨斯世界(post-Malthusian world)的政治发展。
人类社会对制度持强烈的保守态度,不会每过一代就把台面上的赌注一扫而光。新制度往往重叠在既有制度上面,例如分支世系制,它是社会组织最古老的形式,却依然存在于现代世界的很多地方。如不弄清这一遗产和它对今日政治行动者选择的限制,就不可能理解今日改革的可能性。
此外,厘清制度初建时的复杂可帮助我们看到,它们的转变和模仿,即使在现代情形下,也是异常艰难的。政治制度得以建立,往往出于非政治原因(经济学家称之为政治制度的外部因素),我们已看到若干案例。其中之一是私人财产,它的出现不仅为了经济,还因为宗族需要土地埋葬祖先以平息死者灵魂。同样,法治的神圣不可侵犯,在历史上全靠法律的宗教起源。国家在中国和欧洲出现,根源就是当代国际体系所竭力阻止的无休止战争。没有这些外部因素,仍想重建这些制度,往往举步维艰。
我将总结本卷中有关政治制度发展的主题,并从中提炼出政治发展和衰败的理论大纲。这可能算不上真正的预测性理论,因为最终结果往往取决于互有关联的众多因素。此外还有龟的问题,即选来充当原因的龟,结果又要以底下的龟为基础。我以自然状态和人类生物学为本卷的开头,因为它是明显的起点,可算作底层的龟(Grund-Schildkröte),可以背驮后续的龟群。
政治的生物基础
人类在社会中组织自己行为时,不是完全自由的,因为他们共享一种生物本性。考虑到非洲之外的多数当代人,都可认祖归宗到大约五万年前的小群体,这种本性在全世界都是统一的。共享的本性不能决定政治行为,但可限定可能的制度性质。这表示,人类政治取决于人类重复的行为模式,既横跨文化又纵越时间。共享的本性将在下述论点中获得说明:
人类从未在无社会状态中生存。据称,人类曾是隔离的,要么在无政府暴力中与他人互动(霍布斯),要么在和平中对他人一无所知(卢梭),但这却是错的。人类及其灵长目祖先,一直生活在基于亲戚关系的大小社会群体中。生活得如此长久,以至社会合作所需要的认知和情感,都已进化成人类的天性。这表明,有关集体行动的理性选择,即他们核算合作的利弊,大大低估了人类社会既存的合作,也误读了其中的动机。①
人类天生的社会交往建立在两个原则之上:亲戚选择和互惠利他。亲戚选择原则,又称包容适存性原则,是指人类会大致根据共享的基因比例,对跟自己有遗传关系的亲属(或被认为有遗传关系的个体)做出利他行为。互惠利他原则是指,随着与其他个体的长时间互动,人类会发展出共同的利害关系。跟亲戚选择不同,互惠利他不依赖遗传关系,而是依赖重复、直接的人际互动,以及从这类互动中产生的信任关系。在缺乏其他更为非人格化制度激励的情况下,这些形式的社会合作是人类互动的预设模式。一旦非人格化制度出现衰败,这两种合作又会重现,因为这是人类的本性。我所谓的家族化,就是指基于这两项原则的政治用人。所以,当中国汉朝末年皇亲国戚充塞朝廷,当土耳其禁卫军让自己的儿子入伍接班,当法兰西王国卖官鬻爵制造世袭产业,只不过是自然的家族制原则复辟了。
人类天生喜欢制定和遵循规范或规则。从根本上说,制度就是限制个人选择的规则,由此类推,可以说人类天生喜欢建立制度。人们核算如何可获最大私利,从而制定理性规则,与他人一起履行社会契约。人类天生具有认知能力,知道如何解答“囚徒困境”类的合作问题。他们记住过去行为以作未来合作的指南;他们通过闲聊和其他分享,传播和获悉他人的可信度;他们有敏锐的知觉,通过察言观色以侦测谎言和不可信赖的行为;他们掌握分享信息的共同模式,不管是语言的,还是非语言的。在某种意义上,制定和遵循规则是在走捷径,可大大减少社交成本,允许高效率的集体行动。
人类遵循规则的本能,往往基于情感,而非理性。像罪过、可耻、骄傲、愤怒、困窘和赞美,都不是学来的,都不是洛克所谓的出生后、与外界互动时获得的。它们在小孩身上表现得非常自然,小孩依照这基于遗传但寓于文化的规则来组织自己的行为。我们制定和遵循规则的能力很像我们的语言能力:规则的内容是传统的,因社会而异;但规则的“内在结构”和我们的接受能力却是天生的。
人类倾向于将内在价值注入规则,这有助于说明社会的保守和顽固。规则的产生是为了因应特殊情形;之后,情形本身有了变化;久而久之,规则变得过时,甚至严重失调,但社会仍然拽住不放。欧洲人示范了枪械的卓有成效,但马穆鲁克仍予以拒绝,因为他们已向骑士征战注入了特殊情感,这直接导致了他们惨败于应时而变的奥斯曼帝国。因此,各社会都有竭力保留现存制度的普遍倾向。
人类天生具有暴力倾向。从存在的第一瞬间,人类就对其同类行使暴力,就像他们的灵长目祖先。尽管我敬仰卢梭,但暴力倾向不是人类在历史某时某刻学来的。同时,社会制度的存在就是为了控制和转移暴力。政治制度最重要的功能之一就是调控暴力出现的层面。
人类天生追求的不只是物质,还有承认。承认是指对他人尊严或价值的承认,又可称作地位。追求承认或地位的奋斗,往往不同于为物质的奋斗;地位是相对的,不是绝对的,即经济学家罗伯特·弗兰克(Robert Frank)所称的“地位性物品”(positional good)。②换言之,只有他人都处于低级地位时,你才算拥有了高级地位。像自由贸易的合作游戏是正和,允许大家都赢;然而,追求承认或地位的斗争却是零和,你的增益一定是对方的损失。
人类政治活动的大部分都以寻求承认为中心。不管是寻求天命的中国未来君主,打黄巾或赤眉旗号的卑微农民,还是法国红便帽起义军,他们都在追求承认。阿拉伯部落平息相互纠纷,征服北非和中东的大部,这是在为伊斯兰教寻求承认。欧洲战士征服新大陆,打的是基督教的旗帜。近代民主政体的兴起,如避而不谈其内核的平等承认,也是无法理解的。在英国,追求承认的性质循序渐进,从部落或村庄的权利,到英国人民的权利,再到洛克式的天赋人权。
抵制人类只追求物质利益的讲法是很重要的。人类历史中的施暴者,往往不在寻求财富,而在追求承认。冲突的长期持续,远远超过其经济意义。承认有时与财富有关,有时又以财富为牺牲品;如把承认视作另类的“功用”(utility),那就偏于简单,于事无补了。
思想作为原因
在解释社会差异和独特发展路径时,如不把思想当作原因,便无法打造政治发展的理论。在社会科学的术语中,思想是独立的变数;在龟的术语中,思想处在龟群的下层,它的底下绝对没有经济或自然环境的龟。
所有的社会都制造现实的心智模型。这些心智模型在不同因素中——时常是无形的——寻找因果关系,为了使世界更清晰、更可预言、更容易操纵。在早期社会,这些无形因素是精神、魔鬼、上帝、自然,时至今日则演变成抽象概念,像地心吸力、辐射、经济自利、社会阶级等。所有的宗教信仰都是现实的心智模型,都把观察到的现象归因于无法或很难观察的力量。至少从大卫·休谟起,我们懂得,单靠实证资料是无法核实因果关系的。随着现代自然科学的发展,我们改用新的因果理论以控制实验或统计分析,至少可以证伪。有了测试因果的更好办法,人类得以更有效地操纵环境。例如,改用肥料和灌溉来增加粮食产量,而不是牺牲者的血液。每个已知的人类社会都制造现实的心智模型。这表明,这种能力是天生的而不是后学的。
共享的心智模型,尤其是宗教,在促进大规模集体行动方面是至关重要的。建立在理性自利上的集体行动,解释不了世界上客观存在的社会合作和利他主义。③宗教信仰激发人们所做的事,只对财富感兴趣的人通常是不做的,就像我们看到的伊斯兰教 7 世纪在阿拉伯半岛的崛起。信念和文化的分享会增进合作,因为有共同目标,还有应付类似难题的协调。④
很多人看到当代世界的宗教冲突,从而反对宗教,认为它们是暴力和心胸狭隘的来源。⑤这在重叠宗教和多样宗教的世界,可能是千真万确的,但他们忽视了宗教的历史作用。它曾扮演非常重要的角色,允许超越亲友的合作,成为社会关系的来源。此外,世俗的意识形态,如马列主义和民族主义,已在很多当代社会取代宗教信仰,呈现出不相上下的破坏能量,也能激发强烈的信念。
心智模型和规则紧密相连,因为它往往明确指出社会必须遵循的规则。宗教不只是理论,而且是道德规范的处方,要求追随者严格遵守。宗教,就像其颁布的教规,都被注入深厚的情感;信教是为了它的固有价值,不是为了它的准确或有用。宗教信仰,既不能确认,也很难证伪。所有这一切加深了人类社会的保守性。现实的心智模型一经采纳就很难变更,即使出现不利的新证据。
几乎所有已知的人类社会都有某种形式的宗教信仰。这表明,宗教很可能植根于人的天性。就像语言和遵循规则,宗教信仰的内容是传统的,因社会而异,但建立宗教原则的能力却是先天的。⑥我的叙述与宗教的政治影响有关,但不以“宗教基因”的存在与否为前提。即使宗教是后学的,它对政治行为仍施加巨大影响。
像马克思和涂尔干那样的思想家,看到宗教信仰在联合群体上的高效率(或是社区整体,或是阶级整体),从而相信宗教是故意为此打造的。如我们所见,宗教思想与政治经济一起发展,从萨满教(shamanism)和巫术,到祖先崇拜,再到拥有成熟原则的多神论和一神论宗教。⑦宗教信仰与信徒团体的生存条件,必须发生明显的关联。自杀教派,或禁止其成员繁衍的教派,如震教徒(Shakers),就不会存活太久。所以很容易产生一种倾向,以物质条件来解说宗教,并视宗教为它的产物。
然而,这是一个大错。既存的物质条件永远解释不了宗教。最明显的案例是中国和印度的对照。公元前第一个千年终止时,两个社会的社会结构非常相似,都有父系血统的家族和由此产生的政治模式。之后,印度社会转入弯路,唯一的解释是婆罗门宗教的兴起。该教形而上学的主张是非常复杂的,但要把它与当时印度北部的经济和环境条件挂起钩来,却是徒劳无益的。
我描绘的众多案例中,宗教思想都在塑造政治结果方面扮演了独立角色。例如,在欧洲两个重要制度的形成中,天主教会曾发挥主要作用。6 世纪以来,日耳曼野蛮部落逐渐征服罗马帝国;但在颠覆日耳曼的亲戚团体产权结构上,天主教会是关键,更削弱了部落制本身。欧洲由此走出基于亲戚关系的社会组织,用的是社会手段,而不是政治手段,与中国、印度和中东截然不同。在 11 世纪,天主教会宣告独立自主,不受世俗政府的管辖,并将自己组织成现代的等级制度,推动全欧洲的法治。相似的独立宗教机构,也存在于在印度、中东和拜占庭帝国,但在促使独立法律的制度化上,都比不上西方教会。没有叙任权斗争及其后果,法治绝不可能在西方落地生根。
没有案例显示,宗教价值是超越物质利益的。像印度的婆罗门和穆斯林社会的乌里玛,天主教会也是拥有物质利益的社会团体。教宗格里高利一世所颁布的遗产新法,似乎不是为了教义,而是为了私利;它鼓励把土地转让给教会,而不必留给亲戚团体。尽管如此,教会不只是简单的政治参与者,像当时支配欧洲的各式军阀。它无法将资源转换成军事力量,没有世俗政府的帮助,也无法从事掠夺。另一方面,它却可将合法性授予世俗的政治参与者。这件事,后者光凭自己是做不到的。经济学家有时谈起政治参与者如何“投资”于合法性,好像合法性是生产工具,像土地或机器。⑧如要理解合法性,就一定要投入它的特殊语境,即人们对上帝、正义、人生、社会、财富、美德等的观念。
人类价值和意识形态的最重要变化之一——平等的承认——发生于本卷所涵盖时期的结尾,可以说正是这一观念定义了现代世界。人类平等思想有很深的根源:学者如黑格尔、托克维尔和尼采,把现代的平等思想追溯到圣经中以上帝形象造人的说法。然而,享有同等尊严的人类小圆圈,其扩张速度是非常缓慢的,要到 17 世纪之后,才开始包括社会较低阶层、女性、种族、宗教等少数人群体等。
从族团和部落层次的社会迈入国家层次的社会,在某种意义上,代表人类自由的一大挫折。与基于亲戚关系的前任相比,国家更为富饶,更为强大。但这财富和力量,却铸造了悬殊的等级差别,有的变成主人,更多的变成奴隶。黑格尔会说,在如此不平等的社会中,统治者获得的承认是有缺陷的,最终连自己也不满意,因为它来自缺乏尊严的人。现代民主的兴起为所有人提供自决机会,以承认相互的尊严和权利为基础。因此,它只是在更大更复杂的社会里,恢复当初迈入国家时所失去的。
负责制政府出现,与相关思想的传播是分不开的。我们在英国议会的案例中看到,对英国人民权利的信仰是议会团结的根本,洛克式的天赋人权塑造了光荣革命。这些思想进而推动美国革命。我在此书呈现的负责制兴起的历史原因,似乎植根于政治参与者的物质利益,但我们必须同时考虑,确定政治参与者和集体行动范围的相关思想。
政治发展的普遍机制
政治制度的进化,大致可与生物进化媲美。达尔文的进化论以两项简单的原则为基石:变异和选择。有机体的变异,源于基因的随意组合;能更好适应环境的变种,则获得较大的繁殖成功,适应力较差的就要付出代价。
以长远的历史观点看,政治发展遵照同一模式:不同人类群体所使用的政治组织发生了变异,较为成功的——能发展较强的军事和经济力量——得以取代较不成功的。在高层次的抽象中,很难想象政治发展还有他路可走。但先要弄清政治进化与生物进化的差异,其至少有三条。
首先,在政治进化中,选择对象是体现在制度身上的规则,而生物进化中的选择对象是基因。尽管人的天性促进规则的制订和遵循,但不能决定其内容,所以会有内容上的极大差别。制度以规则为基础,将优势授予其生存的社会;在人类代理人的互动中,获选的是优势制度,淘汰的是劣势制度。
其次,在人类社会中,制度的变异可按计划,可作商讨,不像基因变异那样随意。哈耶克强烈驳斥人类社会自觉设计制度的想法,将之追溯到后笛卡尔(post-Cartesian)的理性主义。⑨他认为,社会中多数信息其实是本地的,无法获得中央代理人的理解。⑩哈耶克论点的缺陷是,人类一直在社会各层次成功地设计制度。他不喜欢自上而下、集中的国家社会工程,但愿意接受自下向上、分散的制度革新,尽管后者仍是人为设计的。大规模设计的成功频率,可能低于小规模的,但确有发生。人类很难将意外结果和信息残缺纳入计划,但能作计划的事实表明,自觉建立的制度之变异,比简单的随机而变更有可能适应解决问题。不过,哈耶克仍是正确的,制度进化并不取决于人们设计制度的能力,单是变异和选择,便可取得适时应务的进化结果。⑪
最后,被选择的特征——政治进化中是制度,生物进化中是基因——借文化而获得传递,不靠遗传。就适应性而言,这既是优势,又是劣势。文化特征,如规范、习惯、法律、信念或价值,至少在理论上,可在一代人的时间获得迅速修改,如 7 世纪的伊斯兰教,或 16 世纪丹麦农民的扫盲。另一方面,人们偏爱将内在价值注入心智模型和由此建立的制度,导致制度的经久不衰。相比之下,生物有机体不会敬畏或膜拜自己的基因,如不能帮助生物的存活和繁殖,选择原则便会无情将之去掉。所以,制度进化既可快于也可慢于生物进化。
与生物进化不同,制度可通过模仿而获得扩散。衰弱制度的社会,被强大制度的社会打败了,或干脆消灭了,但也有采纳“防御性现代化”的⑫,从而引进竞争者的制度。17—19 世纪的日本德川幕府时期,治国的封建君主们从葡萄牙人和其他旅客处,很早就获悉火器的存在。但他们正处于长期的军火自我管制中,大家同意不引进火器,因为不想放弃传统的冷兵器战争形式。当美国海军准将马休·佩里(Matthew Perry)和他的“黑船”在 1853 年的东京湾露脸时,执政的精英知道,如果不想成为第二个中国,他们必须终止这种舒适的自我管制来取得美国人所拥有的军事技术。1868 年的明治维新之后,日本引进的不只是军火,还有新式政府、中央官僚体制、新教育制度和其他一系列制度,均借鉴于欧洲和美国。
生物进化既是特别的,又是普遍的。特别进化是指物种适应了特殊环境,并作调整,如著名的达尔文雀(Finches)。普遍进化是指成功的物种跨越本地环境,而向外扩散。所以有大规模的普遍进化,从单细胞到多细胞的有机体,从无性繁殖到有性繁殖,从恐龙到哺乳动物等。政治发展也是如此。行为意义上的现代人类,大约在五万年前离开非洲,迁移到世界各地。他们努力适应遇上的不同环境,开发了不同的语言、文化和制度。同时,某些社会凑巧碰上能提供优势的社会组织。于是,也发生了普遍进化,从族团层次,转到部落层次,再转到国家层次的社会。国家层次社会中,组织较为完善的又击败或吸收组织较差的,使自己的社会组织获得传播和扩散。所以在政治制度的演化中,既有分流,也有汇集。
跟生物进化一样,竞争对政治发展至关重要。如没有竞争,就不会有对制度的选择压力,也不会有对制度革新、借鉴、改革的激励。导致制度革新的最重要竞争之一是暴力和战争。经济生产力的增长,使族团层次向部落层次的过渡得以实现,但直接动机则来自部落社会动员人力的优势。第 5 章中,我讨论了国家原生形成的不同理论,包括经济自利、灌溉、密集人口、地理界限、宗教权威、暴力。虽然,所有因素都发挥了作用;但从自由的部落社会到专制的国家社会,此项艰难的过渡,更像是由保全生命而不是经济利益的需要促成的。浏览诸如中国、印度、中东和欧洲等地国家形成的历史记录,我们看到暴力再一次成了主角。它鼓励国家形成,还鼓励与现代国家相关的特别制度的建立。本章后面还会讲到,合作中遇到的某种问题,除了暴力,没有其他方法。
处处是拱肩
生物学家史蒂芬·古尔德(Stephn Gould)和理查德·列万廷(Richard Lewontin),在 1979 年的文章中,以建筑学上的拱肩(spandrel)来解释生物变异中的不可预知。⑬拱肩是支撑圆屋顶的拱门背线与相邻直角形成的弧形区域。它不是建筑师故意设计的,而是其他精心计划的零件组装后留下的副产品。尽管如此,拱肩开始获得装饰,并随时间的推移而自成一格。古尔德和列万廷主张,有机体身上为某个原因而进化的生物特征,到后来,却能为完全不同的原因,提供适应的优势。

我们在政治进化中看到不少类似拱肩的东西。公司——一个有着与其组成人员清晰可分的身份并可以永久存在的机构——最初是作为宗教组织出现的,没有任何商业目的。⑭天主教会支持女子的继承权,不是想增加女子权利——这在 7 世纪是不合时宜的——而是看上了强大家族手中的珍贵地产,认为这是很好的途径。如果说,教会领袖当时就预见,这将影响亲戚关系的整体,这是很可疑的。最后,忙于叙任权斗争的人,脑海中并没有浮现以独立司法限制政府的想法。当时,那只是一场道德和政治的斗争,为了争取天主教会的独立自主。然而在西方,宗教组织赢得的独立自主,经过长期进化,变成了司法部门的独立自主。法律的宗教基础被世俗来源所取代,但它的结构仍保持原样。所以说,法治本身就是一种拱肩。
实际上,不同制度的历史根源,往往是一长列历史意外事件的产品,没人能够预测。这看起来令人泄气,因为当代社会无法经历同样事件来获得类似制度。但这忽略了政治发展中拱肩的作用,与制度的历史来源相比,制度的功能更为重要。一旦发现,其他社会可以完全出乎意料的方式来模仿和采纳。
制度(机构)⑮
在本卷中,我一直使用亨廷顿对制度的定义,即“稳定、有价值、重复的行为模式”。⑯至于被称作国家的那个制度或机构(the institution called the state),我不仅使用韦伯的定义(在界定的领土上合法行使垄断暴力的组织),还使用他对现代国家的标准(按专门技术和技能合理地分工;使用非人格化的用人制度,对公民行使非人格化的权威)。非人格化的现代国家,不管是建立还是维持,都很困难。家族化——基于亲戚关系和互惠利他的政治用人——是社会关系的自然形式,如果没有其他的规范和鼓励,人类就会回归。
现代组织还有其他特征。亨廷顿列出四条标准来测量国家制度(机构)的发展程度:适应和僵硬,复杂和简单,自主和从属,凝聚和松散。⑰这是指越善于适应、越复杂、越能自主和越凝聚的机构,其发展程度就越是成熟。善于适应的组织,可评估不断变化的外部环境,再修改其内部程序来应对。环境总在变化,所以善于适应的机构活得长久。英国的普通法系统,其法官因应新情形,不断在重新解释和延伸有关法律,就是善于适应的样板。
成熟的机构更为复杂,因为它们有更大的分工和专业化。在酋邦或初期国家中,统治者可能同时又是军事长官、总教士、税务员和最高法院的法官。在高度发达的国家中,这些功能由各自为政的组织承担,它们负有特别使命,需要高度的技术能力。汉朝时期,中国已在中央、郡、地方层次派驻无数官僚机构和部门;虽然比不上现代政府,但与犹如君主家庭简单延伸的早期政府相比,却是一大进步。
自主和凝聚是机构标准的最后两条,如亨廷顿指出的,它们密切相关。自主是指机构开发自觉的集团身份,不受社会其他力量的影响。在第 17 至 19 章讨论法治时,我们看到,法律对政府权力的约束,很大程度上取决于法庭所取得的制度性自治。这里的自治是指不受政治干涉,有权训练、雇用、晋升、惩罚律师和法官。⑱自主与专业化也是紧密相连的,所以,它适宜被看作比较成熟的机构的特征。其他条件都相同的情况下,掌控自身内部升迁的军队,比将军是政治任命的军队,或将军是金钱买来的军队,更具战斗力。
另一方面,凝聚是指政治系统中,不同组织的职责和使命都有明确的界定并被遵从。松散的政治制度中,很多组织参与政府行为,如征税和公共安全,但弄不清到底谁在负责。众多自治机构组成的国家部门,比众多从属机构组成的更有可能是凝聚的。在家族化社会中,领袖的家庭或部落成员,在各政府功能上享有重叠或暧昧的权力,或干脆为特殊个人设立特殊官位。忠诚比公共管理能力更为重要,这种情况迄今仍存在于很多发展中国家(甚至少数发达国家)。国家部门中的官方权力分工,与权力的实际分配不符,导致机构的松散。
制度(机构)的四条标准隐含一个概念,即制度是规则,或是重复的行为模式,比任何掌管机构的个人,都要活得长久。先知穆罕默德,生前以自己的魅力使麦地那部落团结起来,但他没有为阿哈里发的继承留下任何制度。年轻的宗教勉强活过第二代的权力斗争,在很多方面仍在为当初的缺陷付出代价,那就是逊尼派和什叶派的大分裂。穆斯林世界中后来的成功政权,全都依靠制度的创建,像奥斯曼帝国的征募制,招募奴隶军,不依赖个人权力。在中国,皇帝实际上变成属下官僚和繁复规则的囚犯。领袖可塑造机构,而高度发达的机构,不仅比拙劣的领袖活得更长,更有训练和招募优秀领袖的制度。
政治衰败
制度之间的竞争促使政治发展,这是一个动态过程。与此对应的,还有一个政治衰败过程;彼时,社会的制度化越来越弱。政治衰败可在两种形式中发生。制度的建立最初是为了迎接特殊环境的挑战。那环境可以是物质的,如土地、资源、气候和地理,也可以是社会的,如对手、敌人、竞争者和同盟者等。制度一旦形成,倾向于长久存在。如上所述,人类天生偏爱将内在价值注入规则和心智模型。如果没有社会规范、礼仪和其他情感投资,制度便不成其为制度——稳定、富有价值、重复的行为模式。制度长存带有明显的适应价值:如果不存在遵循规则和行为模式的天性,就要不断举行谈判,会给社会稳定带来巨大损失。另一方面,就制度而言,社会是极端保守的;这意味着,促使制度成立的原始条件发生变化时,制度却做不到随机应变。制度与外部环境在变化频率上的脱节,就是政治衰败,就是反制度化。
社会对现存制度的历代投资,导致双重失误:不仅没能调整过时的制度,甚至察觉不到已出毛病。社会心理学家称之为“认知失调”,历史上有无数这样的案例。⑲某社会因优秀制度而变得更富裕,或在军事上更强大,其他竞争力较弱社会的成员,如想继续生存,就必须正确地把上述优势归因于根本性的制度。然而,社会的结果总有多种原因,总能为社会弱点或失败找出似是而非的狡辩。从罗马到中国,众多社会把军事挫折归咎于对宗教的不诚,宁可献上更多的礼仪和牺牲,也不愿全力以赴地重整军队。近代社会里,很容易把社会失败归咎于外国阴谋,不管是犹太人的,还是美帝国主义的,而不愿在自己制度身上寻找原因。
政治衰败的第二种形式是家族制复辟。眷顾家人或互惠的朋友是自然的社会交往,也是人类互动的预设。人类最普遍的政治互动,发生在保护人和依附者之间,领袖以恩惠换取追随者的支持。在政治发展的某些阶段,这种政治组织曾是唯一的形式。但是,随着制度的演化,产生了新的规则,用人标准慢慢改为功能或才干——中国的科举制度、土耳其的征募制、天主教的教士独身制、禁止裙带关系的现代立法。但家族制复辟的压力始终存在。最初以非人格化原因聘入机构的人,仍试图将职位传给自己的孩子或朋友。制度遭受压力时,领袖经常发现自己必须做出让步以保证政治优势,或满足财政需求。
这两种政治衰败,我们可看到很多例子。17 世纪前期,组织良好的满人在北方虎视眈眈,中国的明朝面对与日俱增的军事压力。政权的生存,取决于朝廷能否整顿资源,重建精兵,北上御疆。结果一无所成,因为政府不愿或不能增税。此时,政权与不愿承担更高税赋的精英,处于某种大家都觉满意的共存关系;疏于朝政的皇帝发现,比较容易的对策是让睡着的狗继续睡下去。
家族制复辟是一种循环现象。中国西汉时期建立的非人格化官僚制度,逐渐受到贵族家族的侵蚀;他们试图为自己和后裔在中央政府中保留特权;这些家庭在后来的隋唐两朝仍得以支配中国的官僚机构。埃及的马穆鲁克和土耳其禁卫军先要求成家,再要求自己的孩子进入军事机构,从而破坏了非人格化的奴隶军制度。马穆鲁克一例是对 13 世纪晚期局势的回应,当时蒙古威胁逐渐减退,鼠疫频仍,贸易条件恶化。奥斯曼一例的起因是通货膨胀和预算压力,导致塞利姆一世和苏莱曼一世向土耳其禁卫军做出类似让步。天主教会禁止教士和主教成家以建立现代官僚制度,久而久之也发生故障;神职人员寻求圣职与圣俸的合一,使之成为世袭产业。在法国和西班牙则出现公开的卖官鬻爵,政府部门私有化,再由后裔继承。
这两种政治衰败——制度僵化和家族制复辟——经常同时发生。现存制度的既得利益者,即家族化官僚,会极力阻止改革。如制度彻底崩溃,往往又是他们,凭借其荫庇关系网络出来收拾残局。
暴力和功能失调的均衡
我们除了指出制度长存的自然倾向,还可精确解释制度在适应环境时的姗姗来迟。任何一个制度或制度系统,即便在整体上提供诸如内部和平和产权等的公共服务,也一定会惠顾社会上某些群体,并以其他群体为代价。受惠顾的群体,可能在人身和财产方面感到更加安全,可能因靠近权力而收取租金,可能获得特别的承认和社会地位。这些精英组织在现存制度安排中享有既得利益,会尽力保护现状,除非自我分裂。使全社会获益的制度性变化,如征集土地税以应付外来威胁,仍会遭到组织良好的群体的否决,因为对他们而言,净得仍然是负数。
经济学家很熟悉此种集体行动的失败。博弈理论家称之为稳定的均衡(stable equilibrium),因为没有一名参与者能从现存制度安排的变更中得到个人的好处。但从全社会的角度看,这个均衡是失调的。曼瑟尔·奥尔森认为,任何社会的既得利益群体,经过长年的累积,为保护其狭隘的特权,会组成寻租联合体(rent-seeking coalition)。⑳他们的组织能力远胜过人民大众,所以后者的利益往往在政治制度中得不到代表。失调的政治均衡可借民主而获缓和。民主允许非精英,至少在理论上,获得更多的政治权力。但通常,精英和非精英的组织能力有云泥之别,从而阻止了后者的任何果断行动。
寻租联合体阻止必要的制度变革,从而激发政治衰败;这样的例子不计其数。其经典案例就是法兰西王国,也是租金一词的发源地。其时,法兰西君主在两个世纪中,招诱大部分精英,而逐渐强大。招诱的形式是出卖国家功能的一小部分,之后变成世袭产业。像莫普和杜尔哥这样的改革部长,力图废除卖官鬻爵,却遭到既得利益者强有力的阻挠。卖官问题的解决,最终只有通过法国大革命时期的暴力。
功能失调的均衡(dysfunctional equilibria)很早就出现在人类历史上。考古证据显示,族团层次社会早已掌握农业技术,但持续几代仍坚持狩猎采集。个中原因似乎又是既得利益者。平等的族团层次社会中,分享食物相当普遍,一旦出现农业和私人财产,就难以为继。定居下来的第一户,其生产的粮食必须与族团其他成员分享,反过来摧毁了转向农业的奖励。农业的生产效率,高于狩猎采集。所以,改变生产方式将使全社会更加丰裕,但会剥夺部分成员的免费享用。考古学家斯蒂芬·勒布朗认为,部分狩猎采集社会向农业社会的转变之所以缓慢,就是因为无法解决此类合作问题。㉑
所以,社会能否实施制度变革,取决于能否分化手握否决权的既得利益群体。有时,经济变化削弱现存精英,加强赞成改制的新精英。17 世纪的英国,与商业或制造业相比,地产的回报逐渐降低,从而使资产阶级在政治上获益,吃亏的是旧贵族。有时,新兴的社会参与者因新宗教的涌现而赢得权力,像印度的佛教和耆那教。宗教改革后,由于扫盲和圣经的广泛传播,斯堪的纳维亚的农民不再是毫无生气的一盘散沙。还有的时候,促成变化的是领袖意志和凝聚各无权群体的能力,像叙任权斗争中格里高利七世所组织的教皇派。实际上,这就是政治的精髓:领袖们能否借助权威、合法性、恐吓、谈判、魅力、思想和组织来实现自己的目标。
功能失调的均衡可持续很久,由此说明,暴力为何在制度革新中扮演如此重要的角色。经典的看法认为,政治就是为了解决暴力问题。㉒但有时,要把阻挡制度变革的既得利益者赶走,唯一办法却是暴力。人类对暴毙的害怕强于获利的欲望,由此激发在行为上的深远变化。我们已在第 5 章提到,很难同意经济动机(如实施大型水利工程)是国家原生形成的原因。相比之下,无休止的战争,或害怕较强群体前来征服,促使自由骄傲的部落成员走进集权国家,倒是入情入理的解释。
中国历史上,家族化精英一直是现代国家制度形成的障碍,无论是在秦朝兴起时,还是在隋唐时的复辟期。秦朝方兴的战国时期,贵族带头的无休止战争,摧毁了自己阶层,为非精英军人进入政权打开大门。女皇帝武则天崛起于唐朝早期,清洗传统贵族家庭,促使较为广泛的精英阶层涌现。两次世界大战为 1945 年后走向民主化的德国提供了类似的帮助。它们清除容克贵族阶层(Junker),制度变革遂再无阻挡。
尚不清楚,民主社会能否和平地解决此类难题。美国南北战争之前,南方少数美国人试图竭力保留他们的“特有制度”——奴隶制。只要在美国的西部扩张中,没有足够的自由新州加入以推翻南方的否决权,当时的宪法规则就允许奴隶制的存在。最终,冲突无法在宪政框架内得到解决,战争遂成为必须的选择,六十多万美国人因此而丧生。
现代世界的规范和制度,在很多方面,已把暴力解决政治僵局的大门紧紧关上。没人期望或希望,非洲撒哈拉以南的国家为建立强大巩固的国家,也经历如中国和欧洲所体验的数世纪坎坷。这意味着,制度革新的责任将落在前述的非暴力机制上。不然,社会仍将遇上政治衰败。 幸运的是,国家、法治、负责制这三大基本政治制度得以锻造成功的旧世界,十分不同于当代世界。美国和法国革命以来的两个多世纪,世界经历了工业革命和大幅提高社会交往的技术革新。如今,政治、经济和社会三大组件在发展中的互动,大大不同于 1806 年之前。怎样的互动呢?那是本卷最后一章的主题。
第 30 章 政治发展的过去和现在
自 18 世纪以来,政治发展的条件发生剧烈变化;发展中的政治、经济、社会三个方面,及其在马尔萨斯式世界中的互动;在今天的互动;当代世界的期望
亨廷顿在 1968 年发表《变化社会中的政治秩序》。他的中心见解是,政治发展有其独特逻辑,与经济和社会的逻辑既有关联又有差异。他认为,经济和社会的现代化一旦超越政治发展,政治衰败就会发生,因为现存政治制度无法容纳动员起来的新兴社会群体。他还认为,20 世纪 50 年代和 60 年代独立的发展中国家,之所以遭遇此起彼伏的政变、革命、内战,原因就在这里。
有人认为,政治发展遵循自己的逻辑,未必是整体发展过程中的一部分,看待这个观点要以经典现代化理论为背景。该理论来自 19 世纪的思想家,如马克思、涂尔干、滕尼斯和韦伯。他们试图分析欧洲社会的工业化所引起的巨变。尽管彼此之间有很大差异,他们都倾向于主张,现代化是个整体,包括资本主义市场经济、随之而生的大规模分工、强大的集权官僚国家、亲密的村庄群体变为不近人情的城市群体、公共的社会关系变为个人的社会关系。所有这一切,在马克思和恩格斯的《共产党宣言》中汇聚。该宣言宣称,“资产阶级的兴起”改变了一切,包括劳动条件、全球竞争、最为私密的家庭关系。根据经典现代化理论,这些变化始于 16 世纪早期的宗教改革,在之后三个世纪得到迅猛的展开和传播。
第二次世界大战前,现代化理论家移军美国,抢占地盘,像哈佛大学的比较政治系、麻省理工的国际研究中心、社会科学研究会的比较政治委员会。哈佛大学的比较政治系,由韦伯心爱的学生塔尔科特·帕森斯(Talcott Parsons)领军,希望建立跨学科的社会综合科学,将经济学、社会学、政治学、人类学冶于一炉。①现代化理论家将强烈的规范化价值注入现代化本身,在他们眼中,现代化的好处总会一同到来。经济发展,亲戚团体瓦解,个人主义兴起,更高更包容的教育,价值观以“成就”和理性为方向发生规范性转变,世俗化,民主政治制度的发展;这一切被视为一个相互依赖的整体。经济发展将提供更好教育,导致价值观的改变,依次再促进现代政治,等等,从而取得无止境的良性循环。②
亨廷顿的《变化社会中的政治秩序》,在摧毁现代化理论方面起了重要作用。它强调,现代性的好处不一定相得益彰。尤其是民主,对政治稳定而言,不一定是好事。亨廷顿讲的政治秩序,相当于我在本书中所论的国家建设。他的发展策略,被称作“威权式过渡”(authoritarian transition),主张政治秩序优先于民主,该书因此而变得名闻遐迩。③这也是土耳其、韩国、中国台湾、印尼所走的道路:先在威权统治下实现经济现代化,再在政治制度上开放民主竞争。
本卷呈现的历史材料确证了亨廷顿的基本见解,即发展中的各方面应分开对待。如我们所见的,中国人早在两千多年前,就创造了韦伯式的现代国家,但没有法治或民主,更不用说个人的社会关系或现代资本主义了。
此外,欧洲的发展又与马克思和韦伯的描述大相径庭。欧洲现代化的萌芽远早于宗教改革。我们曾在第 16 章看到,随着日耳曼野蛮人皈依基督教,脱离基于亲戚关系的社会组织,在中世纪黑暗时代便已开始。到 13 世纪的英国,自由买卖财产的个人权利,包括女性的财产权,已属根深蒂固。天主教会 11 世纪晚期与皇帝的争斗是现代法律秩序的根源。它建立欧洲第一个官僚化组织,以管理教会的内部事务。它一直被当作现代化的障碍而横遭诋毁。但从长远看,在推动现代化的关键问题上,它至少像宗教改革一样重要。
所以,欧洲走向现代化,不是全方位的突飞猛进,而是几乎历时一千五百年的点滴改良。在这特有的次序中,社会中的个人主义可早于资本主义,法治可早于现代国家的形成,封建主义作为地方抵抗中央的顽固堡垒,可成为现代民主的基础。根据马克思主义的观点,封建主义是资产阶级上升之前的发展阶段。但在事实上,它主要是欧洲的独特制度。不能把它说成是经济发展的普遍过程,也不能期望非西方社会遵循相似的发展次序。
然后,我们需要分别对待发展中的政治、经济、社会三个方面,弄清它们作为分立的现象,又是如何相互关联、如何周期性互动的。我们必须弄清此事,因为它们现在的相互关系的性质,与在马尔萨斯式世界的历史条件之下,已然十分不同。
托马斯·马尔萨斯
约在 1800 年后,随着工业革命的出现,世界发生了巨大变化。在那之前,生产力因技术革新而持续增长、进而促进经济发展的美事是靠不住的。事实上,它几乎不存在。
但这并不表示,1800 年之前没有发生过生产力的大幅增长。农业、灌溉、铁犁、印刷机、远航帆船,都提高了人均产值。④例如,公元前第三个千年和第二个千年之间,墨西哥特奥蒂瓦坎(Teotihuacán)的农业生产力因引进玉米新品种而增长两倍。⑤那时所缺乏的是年复一年的生产力和人均产值的稳定增长。我们今天假设,电脑和互联网在五年后将获得巨大改进,这很可能是正确的。而中国西汉的农业技术,即基督诞生后不久,与 19 世纪沦为半殖民地之前的清朝的相比,则相差无几。
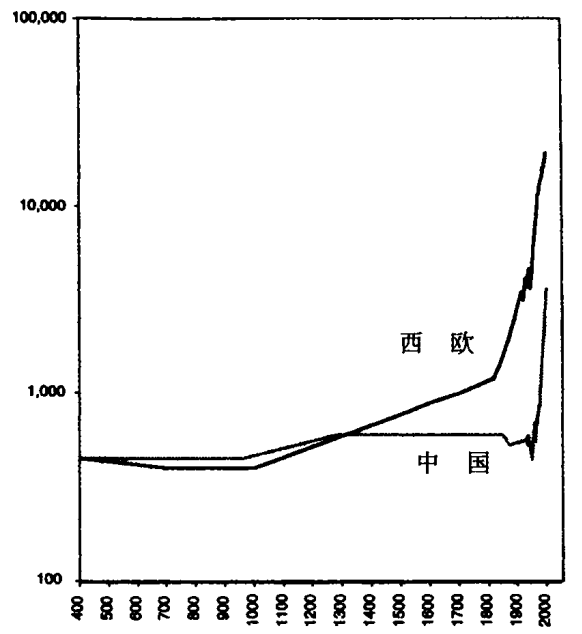
图 7 是西欧和中国在公元 400 年至 2001 年之间的人均产值估计。它显示,从公元 1000 年到 1800 年,西欧的人均收入在八百年期间逐步上升,之后却突飞猛进。同期的中国人均收入,几乎是停滞不前,但在 1978 年后腾飞,速度甚至超过西欧。⑥
1800 年后生产力大幅增长的原因,一直是研究发展的核心命题。首先是智识环境的巨变,促进了现代自然科学、实用科技、复式簿记、专利法和版权的涌现。最后两项又是鼓励不断创新的微观经济制度。⑦注重最近两百年的发展是未可厚非的,但这阻碍了我们对前现代社会政治经济的理解。经济可以持续增长的假设,使我们重视有助于这种增长的制度和条件,如政治稳定、产权、技术和科研。另一方面,如果假设生产力的改进非常有限,社会就会陷入零和的世界,掠夺他人资源往往变成获得权力和财富的更佳途经。
这个生产力低下的世界,因英国神职人员托马斯·马尔萨斯的分析,而引起世人的注意。他的《人口学原理》初版于 1798 年,其时他仅 32 岁。身为八个孩子之一的马尔萨斯认为,人口以几何级数增长(假设女子“自然”生育率是每人生育十五个小孩),而谷物生产以算术级数增长,这表示人均谷物生产只会下降。马尔萨斯还认为,农业效率可以提高,但从长远看,仍跟不上人口增长;实施道德限制,如晚婚和禁欲,可以帮助控制人口的增长(那是避孕尚不普遍的世界);但人口过剩问题的最终解决,还得依靠饥荒、疾病和战争。⑧
马尔萨斯的文章出现于工业革命前夕。如上所述,工业革命引发了 1800 年后生产力的惊人增长,尤其是在开发煤炭和石油的能量上。从 1820 年到 1950 年,全球的能源供应增长六倍,而人口仅增长一倍。⑨随着现代经济世界的出现,马尔萨斯经济学遭到普遍贬斥,譬如说它的眼光短浅,对技术进步过度悲观。⑩但是,如果说马尔萨斯的模式不能用于 1800—2000 年这段时期,它却可作为理解此前世界政治经济的基础。
作为 1800 年前经济生活的一种历史描述,马尔萨斯模式必须作出重要调整。埃斯特·博塞鲁普认为,人口的增加和密集,不仅没有造成饥荒,反而促进了提高效率的技术革新。例如,埃及、美索不达米亚(Mesopotamia)和中国发展出了密集农业模式,实现了大面积灌溉、新高产作物培育和农业工具的改进。⑪因此,人口增长本身未必是件坏事。食物供应量与死亡率并没有直接关联,除非在大饥荒时期。在抑制人口增长上,疾病一直比饥饿更为重要。⑫如食物不够,人类不必死去,可以缩小身躯来降低对卡路里的需求。⑬类似这样的情形似乎就在上一世代的朝鲜发生过,以应付广泛的饥荒。⑭最后,除了人口过剩,本地环境的恶化也是人均谷物生产下降的原因。对人类社会来说,环境破坏不是什么新鲜事(只是今日的规模前所未有)。过去的社会曾杀尽大型动物、侵蚀表土、颠覆当地气候。⑮
经过上述修改的马尔萨斯模式可提供良好架构,帮助我们梳理工业革命前的经济发展。全球人口在过去一万年中有惊人增长,从新石器初期的大约六百万,到 2001 年的六十多亿,这是一千倍的增长。⑯不过,增长的大部发生在 20 世纪;讲得更确切些,在 20 世纪的最后几十年。1820 年之前的经济增长大都是粗放型的,例如,开垦处女地、给沼泽排水、清除森林、填海造地等。新土地一旦得到开发,产量达到当时技术的限度,生活重又回归到零和。一人增加资源,他人必须削减,人均产量得不到持续增长。不管是世界整体还是本地居民,绝对增长之后便是停滞不前和绝对下跌。在全世界范围,人口因疾病而经历大幅度的滑坡。其中一次发生于罗马帝国末期,那时它面对野蛮部落的入侵、饥荒、瘟疫。另一次发生在 13 世纪,蒙古侵占欧洲、中东和中国,并把瘟疫带到世界各地。在 1200 年至 1400 年之间,亚洲人口从大约两亿五千八百万跌至两亿零一百万。在 1340 年至 1400 年之间,欧洲人口从七千四百万跌至五千二百万。⑰
如此缓慢的技术进步具有双刃特性。短期内,它改进生活水平,革新者为此而得益。但较多资源促成人口增加,从而减少人均产量。与革新之前相比,人类平均生活水平并没得到改善。所以,很多历史学家认为,从狩猎采集到农业社会的过渡,反而使人类生活越过越糟。虽然谷物生产的潜力大增,但人类的食谱更为狭窄,从而影响健康。他们为生产粮食消耗更多体力,居住在密集地域,为疾病的蔓延提供温床,等等。⑱
马尔萨斯式世界中的政治
在零和的马尔萨斯世界,人的生存对政治发展有巨大的意义,也与今日的发展大相径庭。马尔萨斯式世界的人们虽有资源,但只有很少的投资机会,譬如促使经济持续增长的工厂、科学研究或教育。如想增加财富,最好走政治途径来从事掠夺,即强夺他人资源。掠夺有两种:享有强制权力的人,可通过征税或赤裸裸的偷窃,夺走社会其他成员的资源;或将社会成员组织起来,去攻击和偷窃邻近社会。为掠夺而组织起来,增强自己的军事或行政能力,往往比投资于生产能力更为有效。
马尔萨斯认识到战争是限制人口的因素,但马尔萨斯的经典模式可能低估了战争在限制人口过剩中的重要性。它作为人口的控制机制,与饥荒和疾病互为表里,因为饥荒和疾病通常由战争引起。跟饥荒和疾病不同,掠夺是一种可以由人有意掌控、用以应付马尔萨斯式压力的手段之一。考古学家斯蒂芬·勒布朗指出,史前社会中的战争和暴力不断,原因就在于人口老是超越环境的支撑能力。换言之,多数人宁可打仗,也不愿挨饿。⑲
马尔萨斯模式加以扩充后,看去就像图 8。像新作物或农具那样的技术进步,会暂时提高人均产量。但假以时日,这个增产会被人口增加或环境破坏所抵消,人均产量再一次降低。阻止贫穷的蔓延有四种主要机制:他们忍饥挨饿,体型变小;死于疾病;从事内部掠夺;向其他社群发起进攻(外部掠夺)。然后,人均产量又会上升,因为土地和粮食变得更为充沛,或因掠夺他人而致富。
在没有持续技术革新的马尔萨斯式世界中,千万不要高估零和思想所占的主导地位。除了掠夺,还有很多大家都可得益的合作机会。农民和城镇居民开展贸易,便可增加大家的福利;政府提倡公共服务,如治安和互相防卫,会使政府本身和百姓都得到好处。事实上,掠夺要求紧密合作,这一事实又是发展政治组织的最重要动机之一。
图 9 表明工业革命之前,马尔萨斯式世界中政治制度与经济发展的关系。集约型经济增长单独处于左上方,没有任何箭头指向它。集约型增长全靠技术进步,但这些进步不可预测,发生时间的前后往往又有很大间隔。对整个制度来说,那时的技术革新是经济学家所谓的外部因素,独立发生,与发展的其他任何方面无关。(博塞鲁普假设,与日俱增的人口密集周期性刺激技术革新,又使技术革新成为内在因素。但它和人口增长之间,又找不到预测或正比的关系。)所以,所发生的经济增长基本上是粗放型的,而不是集约型的。这表示,随着时间的流逝,总体的人口和资源有所增长,但不在人均基础上。
马尔萨斯式世界中至关重要的政治制度是国家,它是取得粗放型经济增长的主要途径。强制能力——军队和警察——是开展外部掠夺(战争和征服)的资源,又可用于国内居民以保障统治者的掌权。反过来,通过征服或征税而获得的资源,又可转换成强制能力。于是,因果关系是双向的。国家提供基本的公共服务,如安全和产权,可提高经济生产力,但仅得一次——即奥尔森所解释的从流寇变成坐寇——但它无法促使生产力持续增长。
国家权力受合法性的影响,法治和社会动员如要影响政治,全凭作为传送带的合法性。在大多数马尔萨斯式社会,合法性以宗教形式出现。中国、拜占庭帝国和其他政教合一的国家,从其控制的宗教权威那里获得合法性。在基于宗教的法治社会中,宗教将合法性赋予独立的法律秩序,后者再向国家颁发或拒绝法律上的批准。
在马尔萨斯社会中动员新的社会群体,会比在当代世界遇上更多限制。在动员惰性社会参与者方面,宗教合法性扮演了很重要的角色,例如 7 世纪的阿拉伯部落和唐朝的佛道两教。罗马帝国时期,基督教在动员新兴精英上发挥了类似作用。在农业社会,宗教经常成为抗议的载体,以反对既有的政治秩序。所以,它不仅能提供合法性,还能制造不稳定。
马尔萨斯式世界中,政治发展的可能性体现在两条主要途径上。第一条围绕国家建设的内部逻辑和粗放型经济增长。政治权力创造经济资源,后者回过来又创造更为强大的政治权力。这个过程自作循环,直到一个极点:对外扩张的政治体遇上物质上的极限,如地理或技术的;或碰上另外一个政治体;或两种情形的组合。这就是在中国和欧洲出现的建国和战争的逻辑。
政治变化的第二条途径与合法性有关。它或者建立法治,或者授权给新兴的社会参与者,以影响国家权力。我所谓的印度弯路,其根源就是婆罗门宗教的兴起,它削弱了印度统治者仿照中国方式集中权力的能力。新兴的社会参与者一旦获得宗教授权,既可对国家权力作出贡献,如阿拉伯人;又可约束君主集权的尝试,如英国议会。
在马尔萨斯式世界,变化的来源相对有限。国家建设的过程非常缓慢,在中国和欧洲都持续了好多世纪。它也避不开政治衰败,政体回到低层次的发展阶段,不得不再从头开始。新兴的宗教或意识形态不时出现,但像技术革新一样,有点靠不住,无法向现存制度提供持续的活力。此外,技术限制了人们和思想在世界上的迁徙和传播。中国秦始皇发明国家的消息,从没传到罗马共和国领袖的耳朵。只有佛教穿越喜马拉雅山脉,抵达中国和东亚其他地区,其他制度大多困顿于自己的出生地。基督教欧洲、中东和印度的法律传统都自我发展,很少相互影响。
当代条件下的发展
现在让我们考察一下,发展的不同方面在工业革命开始后如何互动。最重要的变化是持续性集约型经济增长的出现,从而影响了发展的几乎所有方面。粗放型经济增长继续出现,但在促进政治变化上,其重要性远远比不上人均产量。此外,民主加入国家建设和法治的行列,成为政治发展的组件。这在图 10 中获得说明。
这些不同方面在当代世界的客观关联已有实质性的研究,可在下列关系中得到总结。
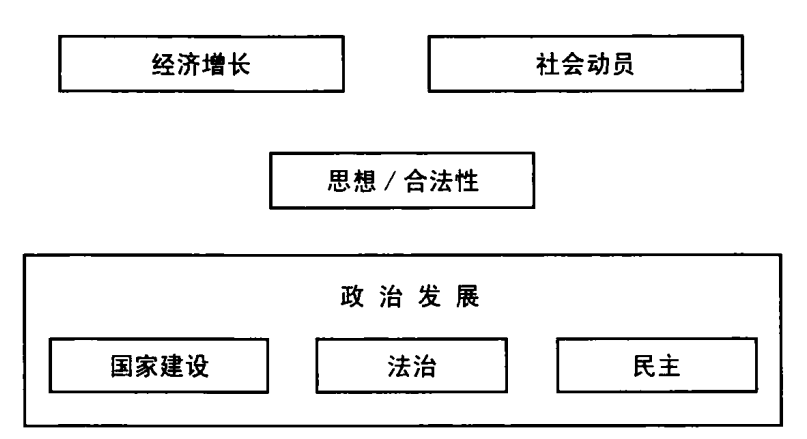
国家建设和经济增长之间
国家是集约型经济增长的基本条件。经济学家保罗·科利尔(Paul Collier)示范了该命题的反面,即国家崩溃、内战、国际冲突对经济增长的负面影响。⑳20 世纪晚期,非洲的大部分贫穷都得归罪于国家的薄弱,以及不时发生的瘫痪和动乱。除了建立国家以提供基本秩序外,强大的行政能力与经济增长呈明显的正相关关系。当人均国内生产总值处于绝对低水平时(少于 1,000 美元),国家变得尤其重要。到了较高水平的收入,国家仍然重要,但其影响可能会发生不成比例的改变。已有很多文献,把良好统治与经济增长联在一起。“良好统治”的定义,因不同作者而各有差异,有时包括政治发展的三大组件。㉑
强大凝聚的国家和经济增长之间的关联早已确定,但相互的因果关系却并不很清楚。经济学家杰弗里·萨克斯(Jeffrey Sachs)认为,良好统治是内生的,不是经济增长的原因,而是它的成果。㉒这听起来很有逻辑,因为政府是大开支。穷国腐败泛滥的原因之一是它们付不起好薪水,以致它们的公务员很难负担家用,所以趋于受贿。政府方面的开支,包括军队、通向学校的道路、街上的警察,在 2008 年的美国大约是人均 17,000 美元,在阿富汗却只有 19 美元。㉓所以一点也不奇怪,阿富汗的国家远远比不上美国,或者,对之大笔援助只会制造腐败。
另一方面,既有经济增长没能促成良好统治的案例,也有良好统治促成经济增长的案例。举韩国和尼日利亚为例。朝鲜战争之后的韩国,1954 年的人均国内生产总值低于尼日利亚,后者在 1960 年从英国手中赢得独立自主。在接下来的五十年中,尼日利亚的石油收入超过三千亿美元。然而,其人均收入却在 1975 年和 1995 年之间出现下跌。相比之下,同期的韩国经济每年增长 7% 到 9%,到 1997 年亚洲金融危机时,已成为世界上第 12 大经济体。这表现上的差异,几乎完全归功于韩国政府,它的治理成绩远远超过尼日利亚。
法治和增长之间
在学术文献中,法治有时被认作统治的组件,有时被认作发展的方面(我在本书的做法)。如第 17 章所指出的,与经济增长有关的法治,涉及产权和合同的强制执行。有大量文献显示,这个关联确实存在。大多数经济学家视之为理所当然,但不清楚,对经济增长来说,普遍和平等的产权是否必不可少。在很多社会中,稳定的产权只为精英而存在,也足以推动经济增长,至少在一段时间内。㉔此外,像当代中国那样的社会,拥有“足够好”的产权,虽然缺乏传统法治,仍能取得很高水平的经济增长。
经济增长和稳定民主之间
社会学家李普塞特(Seymour Martin Lipset)在 20 世纪 50 年代率先注意发展和民主的关联。自那以后,出现了很多将发展与民主连在一起的研究。㉕经济增长和民主之间的关系可能不是线性的——即更多的经济增长并不一定产生更多的民主。经济学家罗伯特·巴罗(Robert Barro)显示,低水平收入时的关联较强,中等水平收入时的关联较弱。㉖有关发展和民主的最完整研究之一显示,从独裁到民主的过渡,可在发展的任何阶段发生,如果人均国内生产总值较高,遇上逆转的机会较小。㉗
经济增长似乎有助于民主的稳定,但逆向的因果关系却不大明显。这似乎很有道理,只要数数近年来取得经济增长惊人纪录的威权政治体——当初仍处于独裁统治的韩国和台湾地区,中国大陆、新加坡、苏哈托(Suharto)治下的印尼、皮诺切特(Augusto Pinochet)治下的智利。因此,凝聚的国家和良好的统治是经济增长的前提,民主是否发挥同样的正面作用,就有点含混不清。
经济增长和社会发展(或公民社会发展)之间
很多古典社会理论将现代公民社会的出现与经济发展联在一起。㉘亚当·斯密在《国富论》中指出,市场增长与社会上的分工有关:市场一旦扩展,公司充分利用规模上的经济效益,社会专业更加精益求精,新兴的社会群体(如工人阶级)得以涌现。现代市场经济所要求的流动性和开放途径,打破了很多传统形式的社会权威,代之以更有弹性的自愿组合。分工愈细所造成的转型效果是 19 世纪思想家著作的中心思想,例如马克思、韦伯和涂尔干。
社会动员和自由民主制之间
自托克维尔开始,大量的民主理论认为,如果没有积极参与的公民社会,现代的自由民主制无法生存。㉙组织起来的社会群体,允许形单影只的个人汇集各自利益,投身政治领域。即使不追求政治目标,志愿组织也会有意外效用,帮助培养在新奇环境中彼此合作的能力——通常被称为社会资本。
上述的经济增长有助于自由民主,恐怕要通过社会动员的途径来生效。经济增长促使社会新参与者出现,随之,他们要求在更为公开的政治制度中获得代表权,从而推动向民主的过渡。如果政治系统已有很好的制度化,便可容纳这些新参与者,然后可有迈向全面民主的成功过渡。这就是 20 世纪的前几十年,随着农民运动和社会党的兴起,在英国和瑞典所发生的。这也是 1987 年军事独裁垮台后,在韩国所发生的。
高度发展的公民社会也能成为民主的危险,甚至可以导致政治衰败。基于民族或种族的沙文主义群体会散播不容忍的偏见;利益群体会尽力追求零和的租金;经济冲突和社会冲突的极度政治化会使社会瘫痪,并破坏民主制度的合法性。㉚社会动员也可导致政治衰败。政治制度拒绝社会新参与者的要求,即所谓的亨廷顿式过程,就发生于 20 世纪 90 年代和 21 世纪初的玻利维亚和厄瓜多尔,高度组织起来的社会群体一再罢免获选的总统。㉛
民主和法治之间
民主的兴起和自由主义法治的兴起在历史上一直有密切关联。㉜如我们在第 27 章所看到的,负责制政府在英国的兴起与保卫普通法不可分。越来越多的公民受到法治保护,这一向被视作民主本身的关键组成部分。这个关联在 1975 年之后的第三波民主化中继续有效,共产主义专政的垮台导致了代议民主制的兴起和立宪政府的建立,以保护个人权利。
思想、合法性和发展的其他方面
有关合法性的思想,其发展有自己的逻辑,但也受经济、政治、社会的发展的影响。如果没有在大英图书馆奋笔疾书的马克思,20 世纪的历史可能会相当不同,他对早期资本主义作了系统性的批判。同样,共产主义在 1989 年的垮台,多半是因为很少人继续信奉马列主义的基本思想。
另一方面,经济和政治的发展影响了人们对思想合法性的认同。对法国人来说,人权的思想顺理成章,因为法国阶级结构已发生变化,还有 18 世纪晚期新兴中产阶级高涨的期待。1929—1931 年的金融大危机和经济受挫,破坏了部分资本主义制度的合法性,使国家干涉经济获得合法性。后来,大福利国家的兴起、经济停滞、由此而生的通货膨胀,为 20 世纪 80 年代保守派的里根—撒切尔(Reagan-Thatcher)革命打下基础。同样,社会主义无法兑现关于现代化和平等的诺言,在共产主义社会的居民眼中,反使自己名誉扫地。
如果政府成功推动经济增长,也可获得合法性。很多迅速发展的东亚国家,如新加坡和马来西亚,即使没有自由民主制,也广受民众支持。相反,经济危机或管理不善所引起的经济倒退,可能动摇政府的稳定,如 1997—1998 年金融危机之后的印尼独裁政府。㉝
合法性也有赖于经济增长的好处分配。如果好处只给处于社会顶端的寡头小集团,没有得到广泛的分享,反而会动员社会群体奋起反对既有的政治制度。这就是波费里奥·迪亚斯(Porfirio Díaz)专政下的墨西哥。从 1876 年到 1880 年,再从 1884 年到 1911 年,他治下的国民收入得到迅速增长,但产权只适用于富裕精英,为 1911 年的墨西哥革命和长期内战创造了条件。其时,弱势群体为争取份内的国民收入而奋斗。最近,委内瑞拉和玻利维亚民主制度的合法性受到民粹领袖的挑战,后者的政治基础是穷人和先前遭到边缘化的群体。㉞
现代发展的范例
发展的不同方面中有多重关联,这表示今天有很多潜在的路径通向现代化,其大部分在马尔萨斯式环境中是无法想象的。让我们以韩国为例,它的发展组件得到特别满意的聚合(参看图 11)。
朝鲜战争结束时,韩国有相对强大的政府。它自中国继承了儒家的国家传统,并在 1905 年到 1945 年的日本殖民期间建成很多现代制度。㉟朴正熙将军 1961 年通过政变上台。韩国在他的领导下,推行工业化政策,以促进经济的迅速增长(箭头 1)。韩国的工业化仅在一代人的时间,就将一个农业穷国改造成为主要的工业强国,并开启了新兴力量的社会大动员——工会、教会团体、大学生和其他传统社会所没有的民间参与者(箭头 2)。全斗焕将军的军政府因 1980 年的光州镇压而丧失合法性,这些新兴的社会团体开始要求军政府下台。在盟友美国的温和推动下,1987 年军政府下台,宣布了总统的首次民选(箭头 3)。经济的迅速增长和国家的民主过渡,帮助加强了政权的合法性。反过来,这又帮助韩国平安渡过 1997—1998 年的严重亚洲金融危机(箭头 4 和 5)。最后,经济增长和民主莅临都有助于韩国法治的加强(箭头 6 和 7)。
在韩国的案例中,如现代化理论所表明的,发展中不同方面倾向于互相支持,彼此加强,尽管有明确的次序,如代议民主制和法治的开始,要等到工业化发生之后。韩国模式未必是普世的,通向现代化还有很多其他路径。在欧洲和美国,法治存在于国家巩固之前。在英国和美国,某种形式的民主负责制早于工业化和经济增长。中国迄今为止走的是韩国路径,但忽略了箭头 3、箭头 4、箭头 7。1978 年邓小平发动经济自由化时,中华人民共和国继承了毛泽东时代相当高效的国家。开放政策促使了未来三十年经济快速增长,数百万农民离开农村,来到城市参加工业就业,社会因此而发生巨大的变迁。经济增长帮助国家取得合法性,并建立公民社会萌芽,但没有动摇政治制度,也没有施加民主化的压力。此外,经济增长导致了法治的改善,因为中国试图将其法律制度提高到世界贸易组织所颁布的标准。中国未来的大问题在于,迅速发展所造成的社会大动员,会导致对更多政治参与的难以抑制的需求。
什么变了
马尔萨斯经济条件下的政治发展和工业革命以来的政治发展,两者的前景如果放在一起考察,可立即看到大量差异,关键是经济持续密集型增长的可能性。人均产量的增长,其所实现的不只是在国家手中注入更多资源。它还刺激社会的广泛转型,动员各式社会新力量,假以时日,将变成政治参与者。相比之下,在马尔萨斯式的世界中,社会动员非常罕见,要是有,大多源于合法性和思想。
传统精英锁在寻租联合体之中,由此造成功能失调的均衡,社会动员是打破这种均衡的重要手段之一。丹麦国王能在 18 世纪 80 年代削弱既得利益的贵族的权力,全靠组织良好的有文化农民的涌现——这是世界历史上的新鲜事,以前只有失序动乱的农民起义。这是工业革命前的社会,动员来源是宗教,打起宗教改革和普遍脱盲的旗号。20 世纪 80 年代,韩国军队和商业精英对权力的掌控,因社会新参与者的出现而被打破。二战后韩国经济起飞时,这些新参与者几乎都尚未问世。政治变化因此而来到丹麦和韩国。丹麦的动员似乎是个意外——丹麦国王选择路德教——而韩国的动员却是马尔萨斯式世界中经济增长的结果,可以预测。在这两件案例中,社会动员在民主传播上都有良好影响,但在其他方面,也导致了政治不稳定。
那时的政治发展与现在相比,另一重要差异是国际因素对国内制度的影响。本书所介绍的几乎所有故事,只涉及单一国家,以及国内不同政治参与者的互动。国际影响基本上是战争、征服、征服的威胁,偶尔还有横跨边界的宗教传播。其时的“跨国”机构,像天主教会和伊斯兰的阿拉伯帝国,在跨越政治边界传播《查士丁尼法典》或伊斯兰教法上,发挥了重要作用。此外,早期现代的欧洲人尝试重新发现古典的希腊和罗马,这属于跨代的学习借鉴。但从整个地球看,发展倾向于各自为政,按地理和地域而分。
今天,情况已有很大不同。我们所谓的全球化,只是数世纪来持续开展过程的最新篇章,其间,与运输、通讯和信息有关的技术在不断蔓延推广。独立发展、几乎没有外界输入的社会,在今天是微乎其微。即使是世界上最隔离最困难的地区,像阿富汗或巴布亚新几内亚,也不能幸免。国际参与者以外国军队、中国伐木公司、世界银行经理的形式崭露头角,不管邀请与否。与以往所熟悉的相比,他们自己也感受到变化的加速。
世界各社会的更大交融增加了互相竞争,其本身就足以制造更频繁的政治变化和政治模式的汇聚。特别进化——即新物种形成和增加生物多样性——发生时,有机体扩散进入明显不同的微型环境,互相之间又失去联系。它的反面是生物全球化,暂栖船舱底层的生物从一个生态区域迁徙到另外一个,可能是意外,也可能是故意。斑马贝、野葛、杀人蜂(Africanized killer bee)都与本土物种展开竞争。这一切,再加上竞争力最强的人类,已导致全球物种数量发生骤减。
这也在政治领域中发生。任何发展中国家可以自由选择自己喜欢的发展模式,无须顾及本土的传统或文化。冷战时期,美国和苏联试图输出各自的政治和经济模式。到了今天,美国仍有促进民主的项目。此外,还有国家指挥的东亚发展模式和中国特色的威权主义。像世界银行、国际货币基金组织和联合国那样的国际机构,随时准备提供关于建立制度的建议,以及资源和技术上的支援,以帮助扩大生产能力。现代的后发达者在制度或政策上无须重新发明车轮。㊱
另一方面,坏事也得以轻易跨越边界——毒品、犯罪、恐怖主义、各式武器、不法资金等。全球化被称作“主权的黄昏”㊲,这未免太夸张。但技术和增长的流动性,使国家很难在自己领土上执法、征税、规范行为、实施与传统政治秩序有关的其他操作。在大多数财富仍体现在土地上的时代,国家可对富裕精英施以相当大的影响。今天,财富可轻易逃至海外的银行账户。㊳
所以已不可能光谈“国家的发展”。在政治学中,比较政治和国际关系,在传统上被认为是明显的分支。前者涉及国内发生的事情,后者涉及国家之间的关系。但现在,这两个领域的研究越来越被当作一个综合体。我们如何到达这一步,政治发展如何在当代世界发生,都将是第 2 卷的主题。
最终,社会并不受困于自己的过去。经济增长、社会新参与者的动员、跨边界社会的组合、竞争和外国模式的流行,都在提供政治变化的契机。在工业革命之前,这些政治变化要么不存在,要么颇受限制。
然而,社会并不能在一代人时间内自由重组自己。全球化对世界各社会的整合,其程度很容易言过其实。社会之间交换和学习的水平远远超越三百年前,但大多数人继续生活在基于传统文化和习惯的环境中。社会惯性仍然很大,外国的制度模式虽比过去更加容易得到,但仍需要融入本土。
必须以恰当的眼光看待本书关于政治制度起源的历史介绍。不应该期望,当代发展中国家必须重蹈中国和欧洲社会所经历的狂暴步骤,以建立现代国家;或现代法治必须以宗教为基础。我们看到,制度只是特殊历史情境和意外事件的产物,不同处境的社会很难予以复制。它们起源的偶然,建立它们所需的持久斗争,应让我们在接受建立当代制度的任务时,备感谦逊。如不考虑现有规则和愿意支持的政治力量,很难将现代制度移植入其他社会。建立制度不像建造水电大坝或公路网络,它需要克服很多困难。首先得说服大家制度变革是必需的;再建立支持者的同盟,以战胜旧制度中既得利益者的抵抗;最后让大家接受新行为准则。通常,正式制度需要新文化的补充。例如,没有独立的新闻界和自我组织的公民社会以监督政府,代议民主制将不会行之有效。
孕育民主的环境和社会条件是欧洲的独特现象,立宪政体似乎因意外事件的环环相连,脱颖而出。但一旦出现,它造就的政治和经济体那么强大,以至在全世界得到广泛的复制。普遍的承认已成为自由民主制的基础,并指向政治发展的初期。其时,社会更加平等,容纳更广泛的参与。我注意到,与取而代之的国家层次社会相比,狩猎采集和部落的社会提供更多的平等和参与。平等尊敬或同等高贵的原则,一旦获得明确的阐述,就很难阻止人们提出此类要求。这可能有助于说明,人人平等的概念在现代世界的无情蔓延,一如托克维尔在《论美国的民主》中所提出的。
今天的负责制
如第 1 章所指出的,民主在世界各地未能得到巩固的原因,与其说是思想本身的呼吁不够,倒不如说是物质和社会条件的缺席,无法促使负责制政府出现。成功的自由民主制,既要有强大统一、能在领土上执法的国家,又要有强大凝聚、能将负责制职责强加于国家的社会。强大国家和强大社会之间的平衡方能使民主生效,不管是在 17 世纪的英国,还是在当代的发达民主国家中。
欧洲早期现代的案例与 21 世纪初的情形之间有很多平行和对照。自第三波开始以来,欲巩固权力的未来威权领袖和希冀民主制度的社会群体,两者之间发生了频繁的斗争。
这是很多苏联继承国的情形,后共产主义的统治者——通常来自前任执政党——开始重建国家,集大权于己身。这也是委内瑞拉、伊朗、卢旺达、埃塞俄比亚的情形。有些地方,像 2000 年之后普京治下的俄罗斯,或 2009 年总统选举之后的伊朗,这种做法得以成功,政治反对派联合不起来,无法阻止专制国家的建立。但在格鲁吉亚和乌克兰,动员起来的政治反对派抵制国家权力,至少在一段时间内获得胜利。在前南斯拉夫,国家彻底崩溃。
早期现代欧洲的情形显然与 21 世纪初大不相同,但仍有集权化和社会抵抗的相似场景。今天有工会、商业团体、学生、非政府组织、宗教组织和其他社会参与者(参看图 12),以取代贵族、士绅、第三等级、农民。当代社会所动员的社会参与者,与我们研究的农业社会相比,更为广泛,更加多样。相关的政治分析,必须弄清国内外不同参与者的性质和凝聚程度。公民社会是否齐心合力和众志成城,或同盟中已有分裂?军队和情报部门是否忠于政权,或存在愿意与反对派谈判的温和路线派?政权的社会基础是什么,掌控怎样的合法性?
今天的国际体系,与我们所研究的早期现代案例相比,对这些斗争有着更大的影响。反对派群体可从国外获得资金、训练、甚至偶尔的武器,而当局也可向志趣相投的盟国呼吁支持。此外,经济全球化提供财政收入的其他来源,如自然资源的出租或外援,从而允许政府避开自己的公民。国王和议会的征税争执不会在石油丰富的国家发生,可能解释了它们中极少民主政体的原因。
未来会怎样
就未来的政治发展而言,我们可提出迄今尚无答案的两个问题。第一个与中国有关。我从一开始就宣称,现代政治制度由强大的国家、法治、负责制所组成。拥有全部三条的西方社会,发展了充满活力的资本主义经济,因而在世界上占主导地位。中国今天在经济上迅速增长,但三条之中只拥有一条,即强大的国家。这样的情境能否长久?没有法治或负责制,中国能否继续维持经济增长,保持政治稳定?经济增长所引发的社会动员,到底是受控于强大的威权国家,还是激起对民主负责制的强烈追求?国家和社会的平衡长期偏向于前者,如此社会能否出现民主?没有西式的产权或人身自由,中国能否拓展科学和技术的前沿?中国能否使用政治权力,以民主法治社会无法学会的方式,继续促进发展?
第二个与自由民主制的未来有关。考虑到政治衰败,在某一历史时刻取得成功的社会不会始终成功。自由民主制今天可能被认为是最合理的政府,但其合法性仰赖自己的表现。而表现又取决于维持恰当的平衡,既要有必要时的强大国家行为,又要有个人自由。后者是民主合法性的基础,并孕育私营经济的增长。现代民主制的缺点有很多,呈现于 21 世纪早期的主要是国家的软弱。当代民主制太容易成为僵局,什么都是硬性规定,无法作出困难的决策,以确保自己经济和政治的长期生存。民主的印度发现,很难整修自己行将崩溃的公共基础设施——道路、机场、供水和排污系统等——因为既得利益者借用法律和选举制度横加阻挠。欧盟的重要成员发现,显然已负担不起自己的社会福利,但无法作出削减。日本累积了发达国家中最高水平的公债,仍然没有采取措施,以消除经济中阻碍未来增长的僵硬。
还有美国,它无法认真处理长期的财政难题,像健康、社会保障、能源等,似乎在政治上日益陷入功能失调的政治均衡。每个人都同意,必须解决长期的财政困境,但消弭赤字而必需的增税或削减开支,仍受到强大利益群体的阻挠。国家制度的设计基于相互制衡,使难题的解决变得尤其困难,加上美国人在意识形态上的僵硬,使之束缚于既定的对策范围。尽管出现这些挑战,美国不太可能重蹈法兰西王国的覆辙,即公职家族化。但它可能也是只有短期的权宜之计,推迟而不是避免最终的危机,像法国政府那样。
现在回头看,制度的最初出现是为了历史上不确定的原因。其中某些存活并得以流传开来,因为它们能满足某种意义上的普遍需求。这就是为何制度在历史上得以互相结合,为何可以提供政治发展的概论。但制度的继续生存也涉及很多意外。一个人口中位年龄在二十几岁的迅速增长的国家,其政治制度卓有成效,但可能不适合三分之一公民已在退休年龄的停滞社会。如果制度无法适应,社会将面临危机或崩溃,可能被迫改用其他制度。不管是非民主政治制度,还是自由民主制度,它都一视同仁。
但有重要理由相信,政治负责制的社会将胜过没有政治负责制的。政治负责制为制度的改善提供和平途径。中国政治制度在王朝时期一直无法解决的问题是“坏皇帝”,像武则天或万历皇帝。英明领导下的威权制度,可能不时地超越自由民主制,因为它可作出快速决定,不受法律和立法机关的挑战。另一方面,如此制度取决于英明领袖的持续出现。如有坏皇帝,不受制衡的政府大权很容易导致灾难。这仍是当代中国问题的关键,其负责制只朝上,不朝下。 我在卷首指出,这里所提供的制度发展的历史介绍,必须对照工业革命后的不同条件。在某种意义上,我已经重新洗过牌,以便直接解说和修正《变化社会中的政治秩序》所提出的问题。工业化发轫后,经济增长和社会动员取得极为迅速的进展,大大改变了政治秩序三个组件的发展前景。这将是我在第 2 卷解说政治发展时所用的架构。
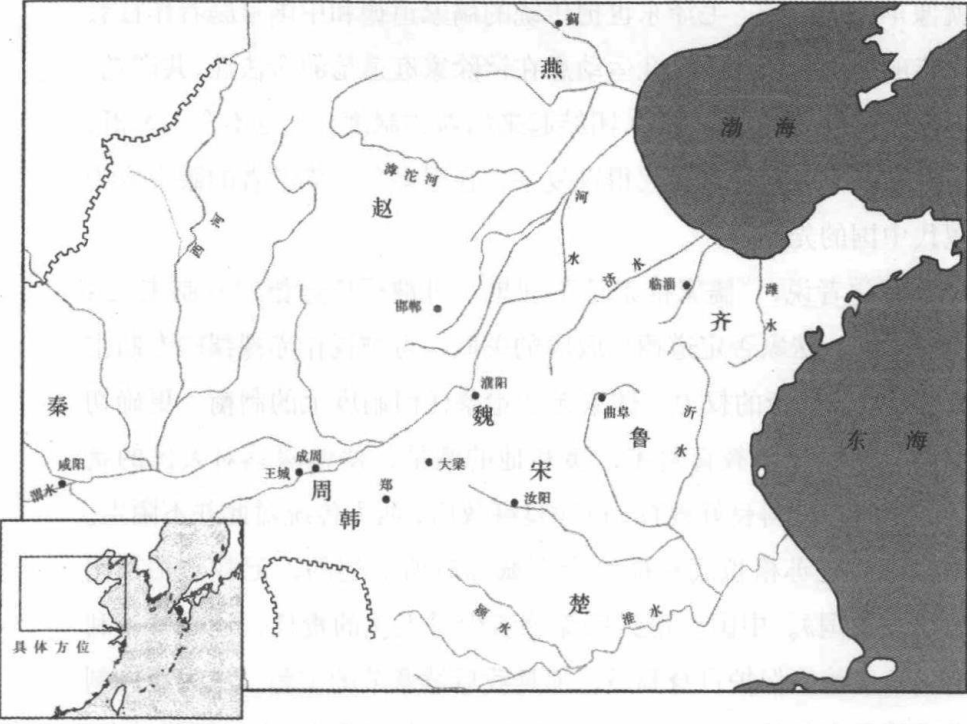
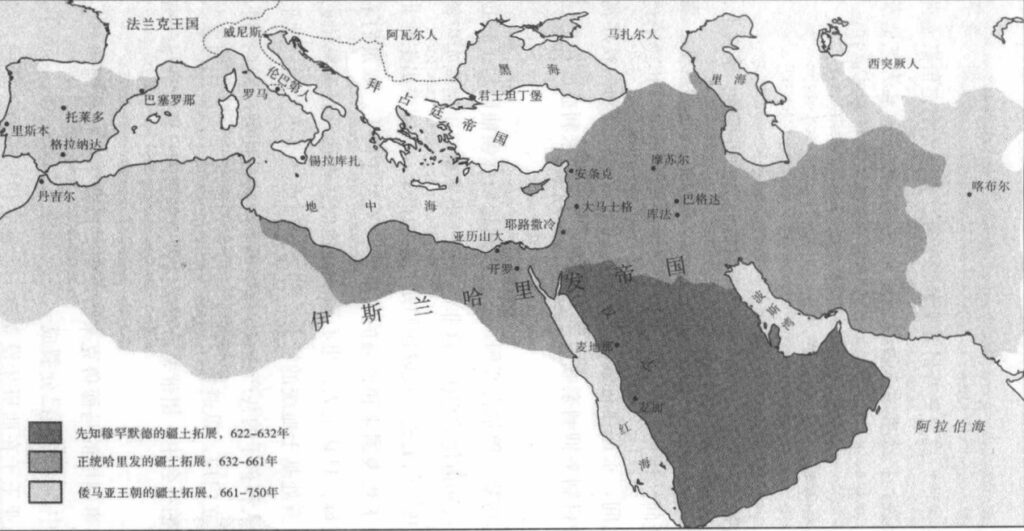

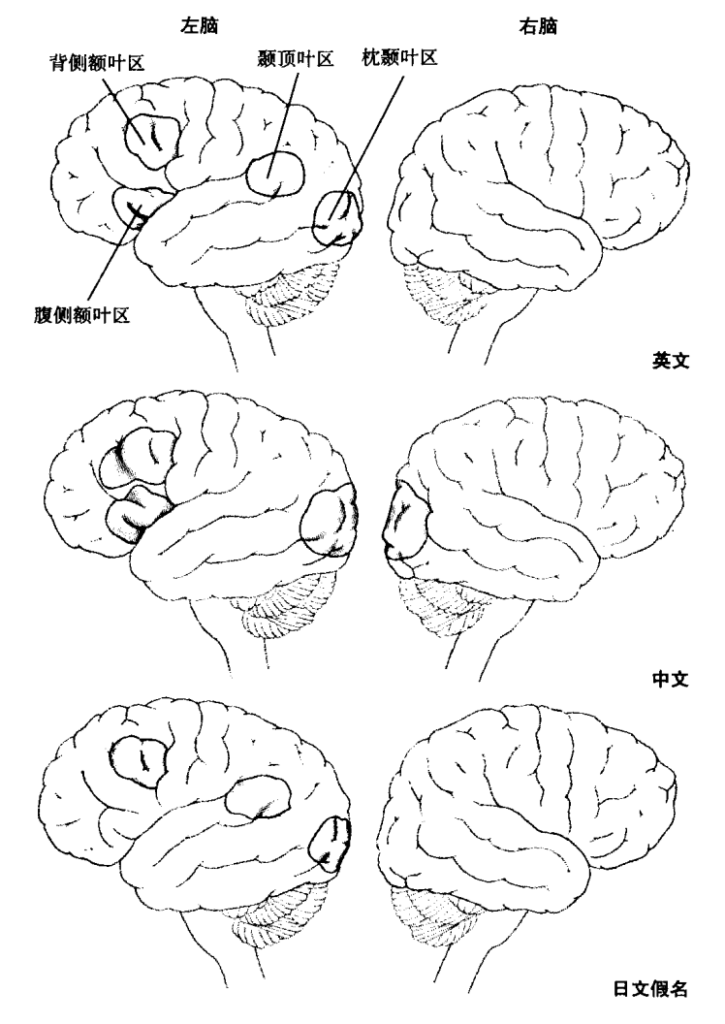


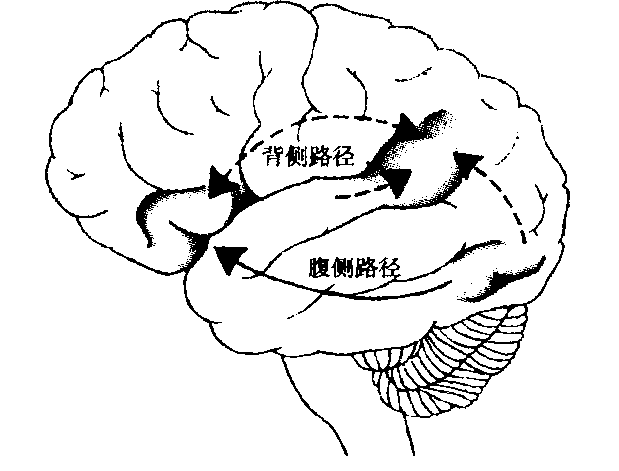
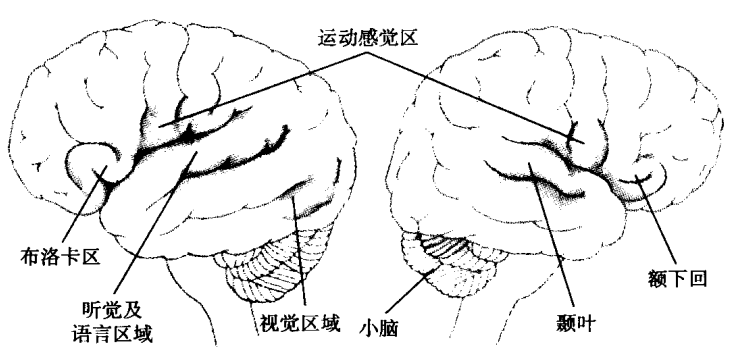
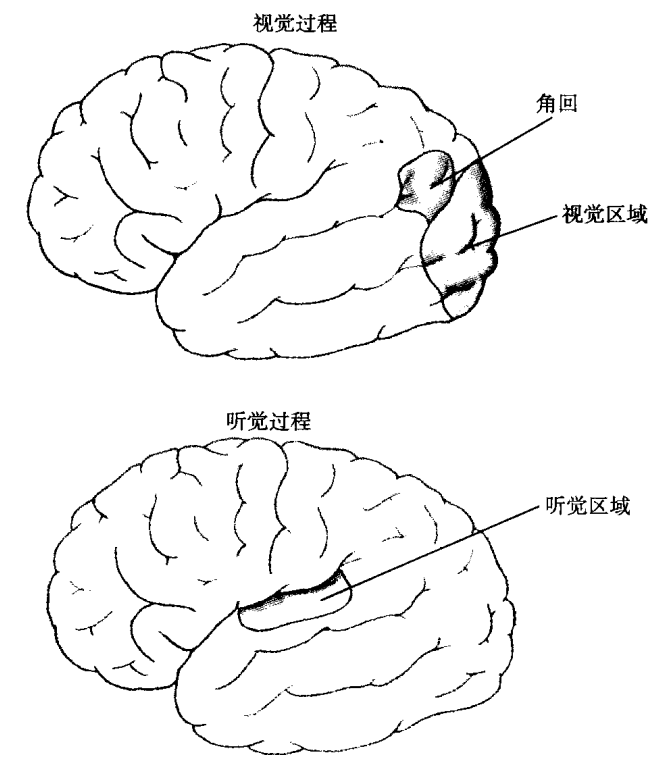
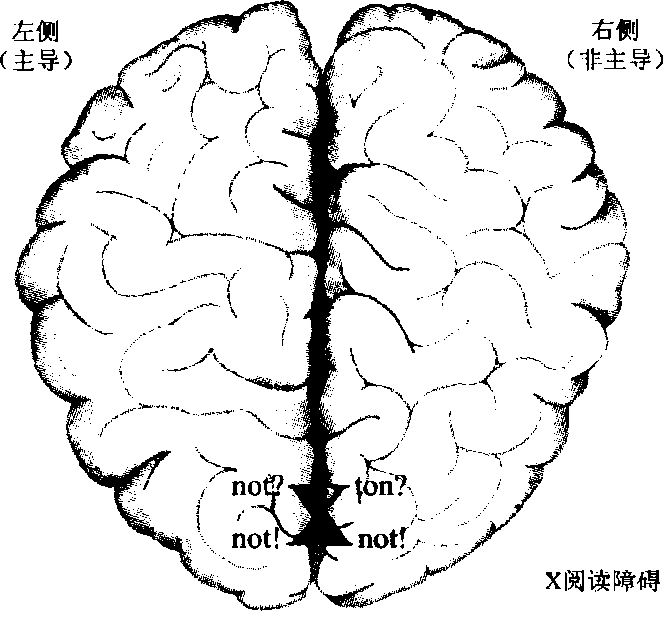
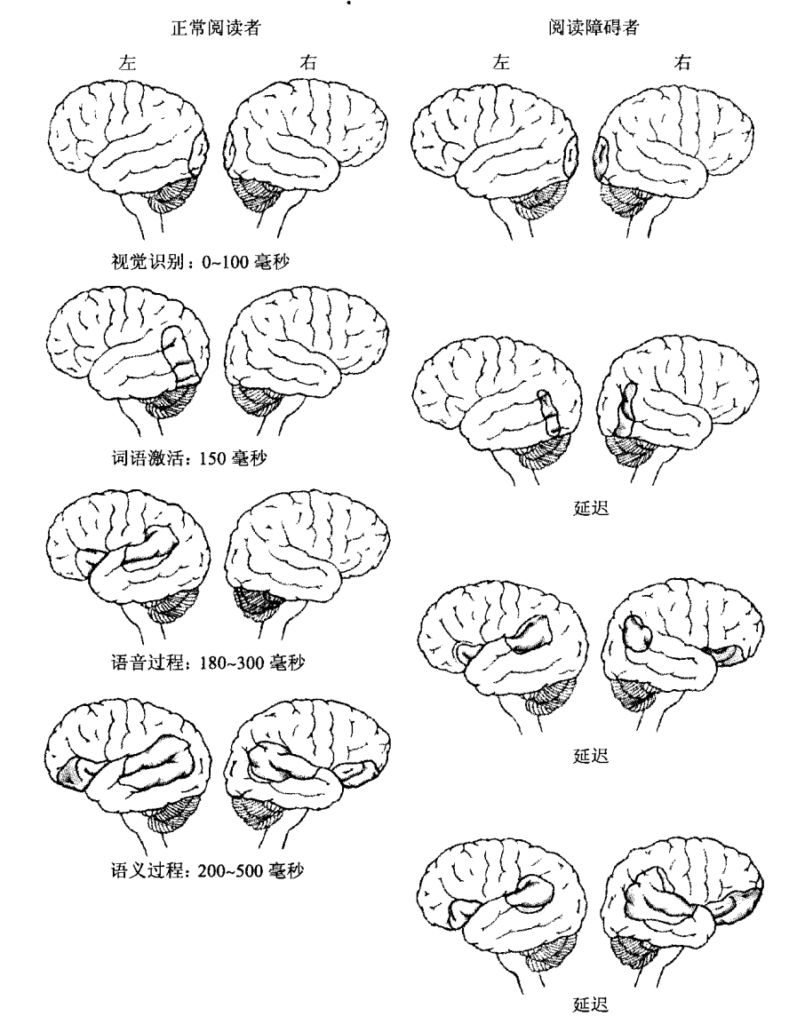

 布各千匹”。至道三年(997年)九月,黎桓遣都知兵马使阮绍恭、副使赵怀德来贡“金银七宝装交椅一、银盆十、犀角象牙五十株、
布各千匹”。至道三年(997年)九月,黎桓遣都知兵马使阮绍恭、副使赵怀德来贡“金银七宝装交椅一、银盆十、犀角象牙五十株、 遣使率部众1600人、马460匹并药物、布帛来贡,宋真宗“厚赏遣还”。
遣使率部众1600人、马460匹并药物、布帛来贡,宋真宗“厚赏遣还”。











